
Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
Epilepsia criptogénica com convulsões em adultos
Médico especialista do artigo

Última revisão: 04.07.2025
De acordo com a classificação internacional em vigor até o ano passado, distinguiam-se entre epilepsia sintomática ou secundária, causada por danos às estruturas cerebrais, idiopática, primária (uma doença independente, presumivelmente hereditária) e criptogênica. Esta última opção significa que os diagnósticos modernos não estabeleceram nenhuma causa para as crises epilépticas periódicas, e a predisposição hereditária também não foi rastreada. O próprio conceito de "criptogênica" é traduzido do grego como "origem desconhecida" (kryptos - secreto, secreto, genos - gerado).
A ciência não para e, talvez, em breve a origem das crises epilépticas periódicas de etiologia desconhecida seja estabelecida. Especialistas sugerem que a epilepsia criptogênica seja uma doença sintomática secundária, cuja gênese não pode ser estabelecida com o nível atual de diagnóstico.
 [
[ Epidemiologia
Epilepsia e síndromes epilépticas são patologias neurológicas muito comuns, que frequentemente levam a consequências graves. A manifestação de crises epilépticas pode ocorrer em pessoas de qualquer sexo e idade. Acredita-se que aproximadamente 5% da população mundial já tenha sofrido pelo menos uma crise epiléptica ao longo da vida.
Anualmente, a epilepsia ou síndrome epiléptica é diagnosticada em uma média de 30 a 50 pessoas em cada 100 mil habitantes da Terra. As crises epilépticas ocorrem com mais frequência em bebês (de 100 a 233 casos por 100 mil pessoas). O pico de manifestação ocorre no período perinatal, após o qual a taxa de incidência cai quase pela metade. As taxas mais baixas ocorrem em pessoas de 25 a 55 anos – cerca de 20 a 30 casos por 100 mil pessoas. Nesse período, a probabilidade de crises epilépticas aumenta e, a partir dos 70 anos, a taxa de incidência é de 150 casos ou mais por 100 mil pessoas.
As causas da epilepsia são estabelecidas em aproximadamente 40% dos casos, portanto, uma doença de etiologia desconhecida não é incomum. Espasmos infantis (síndrome de West), que é uma epilepsia criptogênica, são diagnosticados em crianças de quatro a seis meses de idade, com uma criança com esse diagnóstico ocorrendo em média a cada 3.200 bebês.
Causas epilepsia criptogénica
A base para o diagnóstico da epilepsia são convulsões periódicas, cuja causa é uma descarga elétrica anormalmente forte, que é o resultado da sincronização da atividade das células cerebrais em todas as faixas de frequência, que se expressa externamente no aparecimento de sintomas sensório-motores, neurológicos e mentais.
Para que uma crise epiléptica ocorra, os chamados neurônios epilépticos devem estar presentes, caracterizados pela instabilidade do potencial de repouso (a diferença de potencial entre uma célula não excitada e a membrana externa). Como resultado, o potencial de ação de um neurônio epiléptico excitado tem amplitude, duração e frequência significativamente maiores do que o normal, o que leva ao desenvolvimento de uma crise epiléptica. Acredita-se que as crises epilépticas ocorram em pessoas com predisposição hereditária a tais alterações, ou seja, grupos de neurônios epilépticos capazes de sincronizar sua atividade. Focos epilépticos também se formam em áreas do cérebro com estrutura alterada devido a lesões, infecções, intoxicações e desenvolvimento de tumores.
Assim, em pacientes diagnosticados com epilepsia criptogênica, os métodos modernos de neuroimagem não detectam nenhuma anormalidade na estrutura da massa encefálica, e não há histórico familiar de epilepsia. No entanto, os pacientes apresentam crises epilépticas bastante frequentes, de vários tipos, difíceis de tratar (possivelmente precisamente porque sua causa não é clara).
Assim, os fatores de risco conhecidos para a ocorrência de crises epilépticas – genética, perturbação da estrutura cerebral, processos metabólicos em seus tecidos, consequências de traumatismos cranianos ou processos infecciosos – não são detectados durante exames e pesquisas.
De acordo com a nova classificação de epilepsias de 2017, distinguem-se seis categorias etiológicas da doença. Em vez de sintomática, recomenda-se agora determinar o tipo de epilepsia pela causa estabelecida: estrutural, infecciosa, metabólica, imunológica ou uma combinação delas. A epilepsia idiopática pressupõe a presença de uma predisposição hereditária e agora é chamada de genética. O termo "criptogênico" foi substituído por "fator etiológico desconhecido", o que tornou o significado da formulação mais claro, mas não mudou.
A patogênese da epilepsia é presumivelmente a seguinte: formação de um foco epiléptico, ou seja, uma comunidade de neurônios com eletrogênese prejudicada → criação de sistemas epilépticos no cérebro (com liberação excessiva de mediadores excitatórios, uma “cascata de glutamato” é lançada, afetando todos os novos neurônios e contribuindo para a formação de novos focos de epileptogênese) → formação de conexões interneuronais patológicas → ocorre generalização da epilepsia.
A principal hipótese para o mecanismo de desenvolvimento da epilepsia é a suposição de que o processo patológico é desencadeado por uma violação do estado de equilíbrio entre os neurotransmissores excitatórios (glutamato, aspartato) e aqueles responsáveis pelos processos de inibição (ácido γ-aminobutírico, taurina, glicina, norepinefrina, dopamina, serotonina). O que exatamente viola esse equilíbrio em nosso caso permanece desconhecido. No entanto, como resultado, as membranas celulares dos neurônios sofrem, a cinética dos fluxos iônicos é interrompida - as bombas iônicas são inativadas e, inversamente, os canais iônicos são ativados, a concentração intracelular de íons carregados positivamente de potássio, sódio e cloro é interrompida. A troca iônica patológica através de membranas desestruturadas determina alterações no nível do fluxo sanguíneo cerebral. A disfunção dos receptores de glutamato e a produção de autoanticorpos contra eles causam crises epilépticas. Descargas neurais excessivamente intensas e periódicas, realizadas na forma de crises epilépticas, levam a distúrbios profundos nos processos metabólicos nas células da substância cerebral e provocam o desenvolvimento da próxima crise.
A especificidade desse processo reside na agressividade dos neurônios do foco epiléptico em relação às áreas ainda inalteradas do cérebro, permitindo-lhes subjugar novas áreas. A criação de sistemas epilépticos ocorre no processo de formação de relações patológicas entre o foco epiléptico e os componentes estruturais do cérebro capazes de ativar o mecanismo de desenvolvimento da epilepsia. Tais estruturas incluem: o tálamo, o sistema límbico e a formação reticular da parte média do tronco encefálico. As relações que surgem com o cerebelo, o núcleo caudado do subcórtex e o córtex orbital anterior, por outro lado, retardam o desenvolvimento da epilepsia.
No processo de desenvolvimento da doença, forma-se um sistema patológico fechado – o cérebro epiléptico. Sua formação culmina em um distúrbio do metabolismo celular e da interação de neurotransmissores, circulação cerebral, atrofia crescente dos tecidos e vasos cerebrais e ativação de processos autoimunes cerebrais específicos.
Sintomas epilepsia criptogénica
A principal manifestação clínica desta doença é uma crise epiléptica. Suspeita-se de epilepsia quando o paciente teve pelo menos duas crises epilépticas reflexas (não provocadas), cujas manifestações são muito diversas. Por exemplo, crises semelhantes à epilepsia causadas por febre alta e que não ocorrem em estado normal não são epilepsia.
Pacientes com epilepsia criptogênica podem apresentar convulsões de diferentes tipos e com bastante frequência.
Os primeiros sinais do desenvolvimento da doença (antes do aparecimento de crises epilépticas completas) podem passar despercebidos. O grupo de risco inclui pessoas que sofreram convulsões febris na primeira infância, com a conclusão de que estão mais propensas a convulsões. No período prodrômico, podem ser observados distúrbios do sono, aumento da irritabilidade e labilidade emocional.
Além disso, as crises nem sempre ocorrem na forma generalizada clássica, com quedas, convulsões e perda de consciência.
Às vezes, os únicos sinais iniciais são distúrbios da fala (o paciente está consciente, mas não fala nem responde a perguntas), ou desmaios breves e periódicos. Esses desmaios não duram muito tempo — alguns minutos — e passam despercebidos.
Crises focais simples ou parciais (locais, limitadas) ocorrem com mais frequência, cujas manifestações dependem da localização do foco epiléptico. O paciente não perde a consciência durante o paroxismo.
Durante uma crise motora simples, podem ser observados tiques, espasmos nos membros, cãibras musculares e movimentos rotacionais do tronco e da cabeça. O paciente pode emitir sons inarticulados ou permanecer em silêncio, sem responder a perguntas, estalando os lábios, lambendo-os e realizando movimentos de mastigação.
As crises sensoriais simples são caracterizadas por parestesia – dormência de várias partes do corpo, sensações incomuns de paladar ou olfato, geralmente desagradáveis; distúrbios visuais – flashes de luz, uma grade, manchas diante dos olhos, visão em túnel.
Os paroxismos vegetativos se manifestam por palidez ou hiperemia repentina da pele, aumento da frequência cardíaca, picos de pressão arterial, constrição ou dilatação das pupilas, desconforto na região do estômago até dor e vômitos.
As crises mentais manifestam-se por desrealização/despersonalização e ataques de pânico. Via de regra, são precursoras de crises focais complexas, que já são acompanhadas de comprometimento da consciência. O paciente entende que está tendo uma crise, mas não consegue procurar ajuda. Os eventos que lhe ocorreram durante a crise são apagados da memória do paciente. As funções cognitivas da pessoa são prejudicadas – surge uma sensação de irrealidade do que está acontecendo, e surgem novas mudanças em si mesmo.
Crises focais com generalização subsequente começam simples (complexas), evoluindo para paroxismos tônico-clônicos generalizados. Duram cerca de três minutos e evoluem para sono profundo.
As convulsões generalizadas ocorrem de forma mais grave e são divididas em:
- tônico-clônico, ocorrendo na seguinte sequência: o paciente perde a consciência, cai, seu corpo se curva e se estica em um arco, começam as contrações convulsivas dos músculos por todo o corpo; os olhos do paciente rolam para trás, suas pupilas estão dilatadas neste momento; o paciente grita, fica azul como resultado de parar de respirar por vários segundos, observa-se hipersalivação espumosa (a espuma pode adquirir uma tonalidade rosada devido à presença de sangue, o que indica morder a língua ou a bochecha); às vezes ocorre esvaziamento involuntário da bexiga;
- As crises mioclônicas parecem espasmos musculares intermitentes (rítmicos e arrítmicos) por vários segundos em todo o corpo ou em certas áreas do corpo, que parecem bater os membros, agachar, cerrar os punhos e outros movimentos monótonos; a consciência, especialmente em crises focais, é preservada (esse tipo é mais frequentemente observado na infância);
- ausências - crises não convulsivas com perda de consciência de curto prazo (5 a 20 segundos), expressas no fato de a pessoa congelar com os olhos abertos e inexpressivos e não reagir aos estímulos, geralmente não cai, ao voltar a si, continua a atividade interrompida e não se lembra da crise;
- as ausências atípicas são acompanhadas de quedas, esvaziamento involuntário da bexiga, são mais duradouras e ocorrem nas formas graves da doença, combinadas com retardo mental e outros sintomas de transtornos mentais;
- convulsões atônicas (acinéticas) - o paciente cai bruscamente como resultado da perda do tônus muscular (em epilepsias focais - pode haver atonia de grupos musculares individuais: facial - queda do maxilar inferior, cervical - o paciente senta ou fica em pé com a cabeça baixa), a duração da convulsão não é mais do que um minuto; a atonia nas ausências ocorre gradualmente - o paciente afunda lentamente, em convulsões atônicas isoladas - cai bruscamente.
No período pós-convulsivo, o paciente fica letárgico e inibido; se não for perturbado, ele adormece (especialmente após convulsões generalizadas).
Os tipos de epilepsia correspondem aos tipos de crises. Crises focais (parciais) se desenvolvem em um foco epiléptico local, quando uma descarga anormalmente intensa encontra resistência em áreas vizinhas e se extingue sem se espalhar para outras partes do cérebro. Nesses casos, é diagnosticada epilepsia focal criptogênica.
O curso clínico da doença com foco epiléptico limitado (forma focal) é determinado por sua localização.
Na maioria das vezes, observa-se dano na região temporal. O curso desta forma é progressivo, com convulsões frequentemente mistas, com duração de vários minutos. A epilepsia temporal criptogênica, fora das convulsões, manifesta-se por dores de cabeça, tonturas constantes e náuseas. Pacientes com esta forma de localização queixam-se de micção frequente. Antes da convulsão, os pacientes sentem uma aura precursora.
A lesão pode estar localizada no lobo frontal do cérebro. As convulsões são caracterizadas por uma rapidez repentina, sem aura prodrômica. O paciente apresenta espasmos na cabeça, olhos girando sob a testa e para os lados, e gesticulação automática e bastante complexa é característica. O paciente pode perder a consciência, cair e apresentar espasmos musculares tônico-clônicos por todo o corpo. Com essa localização, observa-se uma série de convulsões de curta duração, às vezes com transição para estado de mal epiléptico generalizado e/ou epilético. Elas podem começar não apenas durante a vigília diurna, mas também durante o sono noturno. A epilepsia frontal criptogênica, em desenvolvimento, causa transtornos mentais (pensamento violento, desrealização) e do sistema nervoso autônomo.
Crises sensoriais (sensação de ar quente se movendo pela pele, toques leves) combinadas com espasmos convulsivos de partes do corpo, distúrbios da fala e motores, atonia, acompanhadas de incontinência urinária.
A localização do foco epiléptico na região orbitofrontal se manifesta por alucinações olfativas, hipersalivação, desconforto epigástrico, além de distúrbios da fala, tosse e edema laríngeo.
Se a hiperatividade elétrica se propagar por todas as partes do cérebro, desenvolve-se uma convulsão generalizada. Nesse caso, o paciente é diagnosticado com epilepsia generalizada criptogênica. Nesse caso, as convulsões são caracterizadas por intensidade, perda de consciência e terminam com o paciente caindo em sono prolongado. Ao acordar, os pacientes queixam-se de dores de cabeça, fenômenos visuais, fadiga e sensação de vazio.
Há também um tipo de epilepsia combinado (quando ocorrem crises focais e generalizadas) e desconhecido.
A epilepsia criptogênica em adultos é considerada, e não sem razão, secundária a um fator etiológico não especificado. Caracteriza-se por convulsões repentinas. Além dos sintomas clínicos, os epilépticos apresentam uma psique instável, temperamento explosivo e tendência à agressividade. A doença geralmente começa com manifestações de alguma forma focal. À medida que a doença progride, as lesões se espalham para outras partes do cérebro; o estágio avançado é caracterizado por degradação pessoal e desvios mentais pronunciados, e o paciente torna-se socialmente desajustado.
A doença tem um curso progressivo e os sintomas clínicos da epilepsia mudam dependendo do estágio de desenvolvimento da epilepsia (grau de prevalência do foco epiléptico).
Complicações e consequências
Mesmo em casos leves de epilepsia focal com crises isoladas e raras, as fibras nervosas são danificadas. A doença tem um curso progressivo, com uma crise aumentando a probabilidade da próxima e a área de dano cerebral se expandindo.
Paroxismos frequentes e generalizados têm um efeito destrutivo no tecido cerebral e podem evoluir para estado de mal epiléptico com alta probabilidade de morte. Há também risco de edema cerebral.
As complicações e consequências dependem do grau de dano às estruturas cerebrais, da gravidade e frequência das convulsões, das doenças concomitantes, da presença de maus hábitos, da idade, da adequação das táticas de tratamento e medidas de reabilitação escolhidas e da atitude responsável em relação ao tratamento do próprio paciente.
Em qualquer idade, lesões de gravidade variável podem ocorrer durante quedas. A hipersalivação e a tendência a engasgar durante uma convulsão aumentam o risco de substâncias líquidas entrarem no sistema respiratório e desenvolver pneumonia por aspiração.
Na infância, há instabilidade no desenvolvimento mental e físico. As capacidades cognitivas frequentemente sofrem.
O estado psicoemocional é instável – as crianças são irritáveis, caprichosas, muitas vezes agressivas ou apáticas, não têm autocontrole e se adaptam mal ao grupo.
Em adultos, esses riscos são agravados por lesões durante a execução de trabalhos que exigem maior atenção. Durante as convulsões, a língua ou a bochecha são mordidas.
Pessoas com epilepsia apresentam maior risco de desenvolver depressão, transtornos mentais e desajustamento social. Pessoas com epilepsia têm limitações na atividade física e na escolha profissional.
Diagnósticos epilepsia criptogénica
No diagnóstico da epilepsia, muitos métodos diferentes são usados para ajudar a diferenciar esta doença de outras patologias neurológicas.
Em primeiro lugar, o médico deve ouvir as queixas do paciente ou de seus pais, se for uma criança. É elaborada uma anamnese da doença – detalhes da manifestação, as especificidades do curso (frequência das crises, desmaios, natureza das convulsões e outras nuances), a duração da doença, a presença de doenças semelhantes em familiares do paciente. Este exame permite presumir o tipo de epilepsia e a localização do foco epiléptico.
Exames de sangue e urina são prescritos para avaliar o estado geral do corpo, a presença de fatores como infecções, intoxicações, distúrbios bioquímicos e para determinar a presença de mutações genéticas no paciente.
Testes neuropsicológicos são realizados para avaliar as capacidades cognitivas e o estado emocional. O monitoramento periódico permite avaliar o impacto da doença no sistema nervoso e na psique, além de ajudar a determinar o tipo de epilepsia.
No entanto, antes de tudo, trata-se de um diagnóstico instrumental, graças ao qual é possível avaliar a intensidade da atividade elétrica das regiões cerebrais (eletroencefalografia), a presença de malformações vasculares, neoplasias, distúrbios metabólicos, etc. em suas regiões.
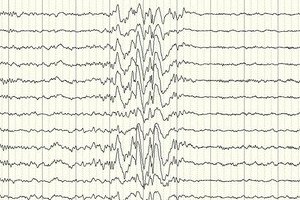
A eletroencefalografia (EEG) é o principal método diagnóstico, pois mostra desvios da norma na intensidade das ondas cerebrais, mesmo fora de um ataque – aumento da prontidão para convulsões em certas áreas ou em todo o cérebro. O padrão de EEG da epilepsia parcial criptogênica é a atividade de espícula-onda ou de onda lenta sustentada em certas partes do cérebro. Usando este estudo, o tipo de epilepsia pode ser determinado com base na especificidade do eletroencefalograma. Por exemplo, a síndrome de West é caracterizada por ondas lentas arrítmicas irregulares, virtualmente não sincronizadas, com amplitude e descargas de espícula anormalmente altas. Na maioria dos casos de síndrome de Lennox-Gastaut, o eletroencefalograma durante a vigília revela atividade de espícula-onda lenta generalizada irregular com uma frequência de 1,5-2,5 Hz, frequentemente com assimetria de amplitude. Durante o repouso noturno, esta síndrome é caracterizada pelo registro de descargas rítmicas rápidas com uma frequência de aproximadamente 10 Hz.
No caso da epilepsia criptogênica, esta é a única maneira de confirmar sua presença. Embora existam casos em que, mesmo imediatamente após uma convulsão, o EEG não registra alterações na forma das ondas cerebrais. Isso pode ser um sinal de que ocorrem alterações na atividade elétrica nas estruturas profundas do cérebro. Alterações no EEG também podem estar presentes em pacientes sem epilepsia.
Métodos modernos de neurovisualização são necessariamente utilizados – computadorizada, ressonância magnética, tomografia por emissão de pósitrons. Este diagnóstico instrumental permite avaliar alterações na estrutura da substância cerebral devido a lesões, anomalias congênitas, doenças, intoxicações, detectar neoplasias, etc. A tomografia por emissão de pósitrons, também chamada de ressonância magnética funcional, ajuda a identificar não apenas distúrbios estruturais, mas também funcionais.
Focos mais profundos de atividade elétrica anormal podem ser detectados pela tomografia computadorizada por emissão de fóton único, e a espectroscopia de ressonância pode detectar distúrbios em processos bioquímicos no tecido cerebral.
Um método diagnóstico experimental e pouco difundido é a magnetoencefalografia, que registra ondas magnéticas emitidas pelos neurônios cerebrais. Ela permite estudar as estruturas mais profundas do cérebro, inacessíveis à eletroencefalografia.
Diagnóstico diferencial
O diagnóstico diferencial é realizado após a realização dos exames mais completos. O diagnóstico de epilepsia criptogênica é feito pela exclusão de outros tipos e causas de crises epilépticas identificadas durante o processo diagnóstico, bem como da predisposição hereditária.
Nem todas as instituições médicas têm o mesmo potencial diagnóstico, portanto, tal diagnóstico requer mais pesquisas diagnósticas em um nível mais alto.
Tratamento epilepsia criptogénica
Não existe um método único para tratar a epilepsia, no entanto, padrões claros foram desenvolvidos e seguidos para melhorar a qualidade do tratamento e a vida dos pacientes.
Prevenção
Como as causas desse tipo específico de epilepsia não foram estabelecidas, as medidas preventivas têm um foco geral. Um estilo de vida saudável – sem maus hábitos, boa alimentação e atividade física – proporciona boa imunidade e previne o desenvolvimento de infecções.
Prestar muita atenção à sua saúde, exames e tratamento oportunos de doenças e lesões também aumentam a probabilidade de evitar essa doença.
Previsão
A epilepsia criptogênica se manifesta em qualquer idade e não apresenta um complexo de sintomas específico, mas se manifesta de forma muito diversa – diferentes tipos de convulsões e síndromes são possíveis. Até o momento, não existe um método único para a cura completa da epilepsia, mas o tratamento antiepiléptico ajuda em 60 a 80% dos casos de todos os tipos de doenças.
Em média, a doença dura 10 anos, após os quais as convulsões podem cessar. No entanto, 20 a 40% dos pacientes sofrem de epilepsia por toda a vida. Cerca de um terço de todos os pacientes com qualquer tipo de epilepsia morrem de causas associadas a ela.
Por exemplo, as formas criptogênicas da síndrome de West têm um prognóstico desfavorável. Na maioria dos casos, evoluem para a síndrome de Lennox-Gastaut, cujas formas leves são passíveis de controle medicamentoso, enquanto as formas generalizadas com convulsões frequentes e graves podem permanecer por toda a vida e ser acompanhadas de grave degradação intelectual.
Em geral, o prognóstico depende muito do momento do início do tratamento; quando ele é iniciado em estágios iniciais, o prognóstico é mais favorável.
A epilepsia pode resultar em incapacidade permanente. Se uma pessoa desenvolver um distúrbio de saúde persistente como resultado da doença, levando à limitação das atividades da vida, isso será determinado por um exame médico e social. Este exame também decidirá sobre a atribuição de um grupo específico de deficiência. Você deve primeiro entrar em contato com seu médico assistente sobre esta questão, que apresentará o paciente à comissão.