
Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
Polimiosite e dermatomiosite: causas, sintomas, diagnóstico, tratamento
Médico especialista do artigo

Última revisão: 05.07.2025
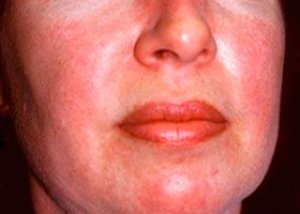
Polimiosite e dermatomiosite são doenças reumáticas sistêmicas raras, caracterizadas por alterações inflamatórias e degenerativas nos músculos (polimiosite) ou nos músculos e na pele (dermatomiosite). A manifestação cutânea mais específica é a erupção heliotrópica.
O envolvimento muscular é simétrico e inclui fraqueza, alguma sensibilidade e subsequente atrofia dos músculos da cintura pélvica proximal. As complicações podem incluir envolvimento visceral e malignidade. O diagnóstico baseia-se na apresentação clínica e na avaliação da disfunção muscular por meio da dosagem dos níveis enzimáticos, realização de ressonância magnética, eletromiografia e biópsia muscular. O tratamento envolve glicocorticoides, às vezes em combinação com imunossupressores ou imunoglobulinas intravenosas.
As mulheres adoecem duas vezes mais que os homens. A doença pode ocorrer em qualquer idade, mas é mais frequentemente detectada na faixa etária de 40 a 60 anos; em crianças, de 5 a 15 anos.
 [
[ O que causa dermatomiosite e polimiosite?
Acredita-se que a causa da doença seja uma reação autoimune ao tecido muscular em indivíduos geneticamente predispostos. A doença é mais comum na presença de histórico familiar grave e portadores de certos antígenos HLA (DR3, DR52, DR56). Possíveis gatilhos são miosite viral e neoplasias malignas. Há relatos da detecção de estruturas semelhantes a picornavírus em células musculares; além disso, vírus podem induzir doenças semelhantes em animais. A associação de tumores malignos com dermatomiosite (muito menos frequente do que com polimiosite) sugere que o crescimento tumoral também pode ser um gatilho para o desenvolvimento da doença, como resultado do início de reações autoimunes a antígenos comuns do tumor e do tecido muscular.
Depósitos de IgM, IgG e do terceiro componente do complemento são encontrados nas paredes dos vasos sanguíneos dos músculos esqueléticos; isso é especialmente característico da dermatomiosite em crianças. Pacientes com polimiosite também podem desenvolver outros processos autoimunes.
Fisiopatologia da dermatomiosite e polimiosite
As alterações patológicas incluem dano celular e atrofia em um contexto de inflamação de gravidade variável. Os músculos das extremidades superiores e inferiores, bem como da face, são menos danificados do que outros músculos esqueléticos. Danos aos músculos viscerais da faringe e do esôfago superior, menos frequentemente do coração, estômago ou intestinos, podem levar à disfunção dos órgãos mencionados. Altas concentrações de mioglobina devido à rabdomiólise podem causar danos renais. Alterações inflamatórias nas articulações e nos pulmões também podem ocorrer, especialmente em pacientes com anticorpos antissintetase.
Sintomas de dermatomiosite e polimiosite
O início da polimiosite pode ser agudo (especialmente em crianças) ou subagudo (mais frequentemente em adultos). A infecção viral aguda às vezes precede ou desencadeia a manifestação da doença, cujas manifestações mais comuns são fraqueza dos músculos proximais ou erupções cutâneas. A dor se manifesta em menor grau do que a fraqueza. Poliartralgia, fenômeno de Raynaud, disfagia, distúrbios pulmonares e sintomas gerais (aumento da temperatura corporal, perda de peso, fraqueza) podem se desenvolver. O fenômeno de Raynaud é frequentemente encontrado em pacientes com doenças concomitantes do tecido conjuntivo.
A fraqueza muscular pode progredir ao longo de várias semanas ou meses. No entanto, para a manifestação clínica da fraqueza muscular, pelo menos 50% das fibras musculares devem ser afetadas (portanto, a presença de fraqueza muscular indica a progressão da miosite). Os pacientes podem ter dificuldade para levantar os braços acima do nível dos ombros, subir escadas ou levantar-se da posição sentada. Devido à fraqueza grave dos músculos pélvicos e da cintura escapular, os pacientes podem ficar confinados a uma cadeira de rodas ou à cama. Se os flexores do pescoço forem afetados, torna-se impossível levantar a cabeça do travesseiro. O acometimento dos músculos da faringe e do esôfago superior leva a distúrbios de deglutição e regurgitação. Os músculos dos membros inferiores, superiores e da face geralmente não são afetados. No entanto, podem ocorrer contraturas dos membros.
As erupções cutâneas observadas na dermatomiosite são geralmente escuras e eritematosas. Edema periorbital de coloração roxa (erupção heliotrópica) também é característico. As erupções cutâneas podem ser ligeiramente elevadas acima do nível da pele e ser lisas ou escamosas; a localização das erupções cutâneas é a testa, pescoço, ombros, tórax, costas, antebraços, partes inferiores das canelas, sobrancelhas, áreas dos joelhos, maléolos mediais, superfícies dorsais das articulações interfalângicas e metacarpofalângicas, na face lateral (sintoma de Gottron). Hiperemia da base ou da periferia das unhas é possível. Dermatite descamativa, acompanhada de rachaduras, pode se desenvolver na pele da superfície lateral dos dedos. Lesões cutâneas primárias geralmente se resolvem sem sequelas, mas podem levar a alterações secundárias, como pigmentação escura, atrofia, cicatrizes ou vitiligo. Calcificações subcutâneas podem se desenvolver, especialmente em crianças.
Aproximadamente 30% dos pacientes desenvolvem poliartralgia ou poliartrite, frequentemente acompanhadas de edema e derrame articular. No entanto, a gravidade das manifestações articulares é pequena. Elas ocorrem com mais frequência quando anticorpos contra Jo-1 ou outras sintetases são detectados nos pacientes.
O envolvimento de órgãos internos (exceto faringe e esôfago superior) é menos comum na polimiosite do que em outras doenças reumáticas (particularmente LES e esclerose sistêmica). Raramente, especialmente na síndrome antissintetase, a doença se manifesta como pneumonite intersticial (na forma de dispneia e tosse). Arritmias cardíacas e distúrbios de condução podem se desenvolver, mas geralmente são assintomáticos. Manifestações gastrointestinais são mais comuns em crianças que também apresentam vasculite e podem incluir vômitos com sangue, melena e perfuração intestinal.
Aonde dói?
Classificação da polimiosite
Existem 5 tipos de polimiosite.
- A polimiosite idiopática primária, que pode ocorrer em qualquer idade, não envolve a pele.
- A dermatomiosite idiopática primária é semelhante à polimiosite idiopática primária, mas envolve a pele.
- Polimiosite e dermatomiosite associadas a neoplasias malignas podem ocorrer em pacientes de qualquer idade; seu desenvolvimento é mais frequentemente observado em pacientes idosos, bem como em pacientes com outras doenças do tecido conjuntivo. O desenvolvimento de neoplasias malignas pode ser observado tanto 2 anos antes quanto 2 anos depois do início da miosite.
- A polimiosite ou dermatomiosite infantil está associada à vasculite sistêmica.
- Polimiosite e dermatomiosite também podem ocorrer em pacientes com outras doenças do tecido conjuntivo, mais comumente esclerose sistêmica progressiva, doença mista do tecido conjuntivo e LES.
A inclusão da miosite dos músculos do tronco no grupo da polimiosite é incorreta, visto que esta é uma doença distinta, caracterizada por manifestações clínicas semelhantes às da polimiosite idiopática crônica. No entanto, ela se desenvolve na velhice, frequentemente afeta os músculos das partes distais do corpo (por exemplo, extremidades superiores e inferiores), tem duração mais longa, responde menos ao tratamento e é caracterizada por um quadro histológico típico.
Diagnóstico de dermatomiosite e polimiosite
A polimiosite deve ser suspeitada em pacientes com queixas de fraqueza muscular proximal, com ou sem sensibilidade. A avaliação para dermatomiosite é necessária em pacientes com queixas de erupção cutânea semelhante a heliotrópio ou sinal de Gottron, bem como em pacientes com manifestações de polimiosite combinadas com quaisquer lesões cutâneas consistentes com dermatomiosite. As manifestações clínicas da polimiosite e da dermatomiosite podem assemelhar-se às da esclerose sistêmica ou, menos comumente, do LES ou da vasculite. A certeza do diagnóstico é aumentada pelo atendimento do maior número possível dos cinco critérios a seguir:
- fraqueza dos músculos proximais;
- erupções cutâneas características;
- aumento da atividade das enzimas do tecido muscular (creatina quinase ou, na ausência de aumento de sua atividade, aminotransferases ou aldolase);
- alterações características na miografia ou ressonância magnética;
- alterações histológicas características em uma biópsia de tecido muscular (critério absoluto).
A biópsia muscular pode excluir algumas condições clinicamente semelhantes, como miosite dos músculos do tronco e rabdomiólise devido a infecção viral. As alterações reveladas pelo exame histológico podem variar, mas inflamação crônica e focos de degeneração e regeneração muscular são típicos. Um diagnóstico preciso (geralmente por verificação histológica) é necessário antes de iniciar um tratamento potencialmente tóxico. A ressonância magnética pode detectar focos de edema e inflamação nos músculos, seguidos por uma biópsia direcionada.
Exames laboratoriais podem confirmar ou, ao contrário, afastar a suspeita da presença da doença, sendo também úteis na avaliação da sua gravidade, na possibilidade de associação com outras patologias semelhantes e no diagnóstico de complicações. Apesar de serem detectados anticorpos antinucleares em alguns pacientes, este fenómeno é mais típico de outras doenças do tecido conjuntivo. Cerca de 60% dos pacientes apresentam anticorpos contra o antígeno nuclear (PM-1) ou células totais do timo e contra o Jo-1. O papel dos autoanticorpos na patogénese da doença permanece incerto, embora se saiba que os anticorpos contra o Jo-1 são um marcador específico da síndrome antissintetase, incluindo alveolite fibrosante, fibrose pulmonar, artrite e fenómeno de Raynaud.
A avaliação periódica da atividade da creatina quinase é útil para monitorar o tratamento. No entanto, em pacientes com perda muscular grave, a atividade enzimática pode ser normal, apesar da presença de miosite crônica ativa. A ressonância magnética, a biópsia muscular ou a elevação da atividade da creatina quinase costumam ser úteis na diferenciação entre recidiva de polimiosite e miopatia induzida por glicocorticoides.
Como muitos pacientes apresentam neoplasias malignas não diagnosticadas, alguns autores recomendam o rastreamento de todos os adultos com dermatomiosite e aqueles com polimiosite com mais de 60 anos, utilizando o seguinte esquema: exame físico, incluindo exames de mama, pélvico e retal (incluindo pesquisa de sangue oculto nas fezes); hemograma completo; bioquímica sanguínea; mamografia; teste de antígeno carcinoembrionário; urinálise; radiografia de tórax. Alguns autores questionam a necessidade desse rastreamento em pacientes mais jovens que não apresentam evidências clínicas de malignidade.
O que precisa examinar?
Tratamento de dermatomiosite e polimiosite
Até que a inflamação seja aliviada, a atividade física deve ser limitada. Os glicocorticoides são os medicamentos de primeira linha. Na fase aguda da doença, os pacientes adultos devem receber prednisolona (por via oral) na dose de 40 a 60 mg por dia. A determinação regular da atividade da creatina quinase é um indicador precoce de eficácia: na maioria dos pacientes, observa-se uma diminuição ou normalização dentro de 6 a 12 semanas após um aumento na força muscular. Após a normalização da atividade enzimática, a dose de prednisolona é reduzida: primeiro em aproximadamente 2,5 mg por dia durante uma semana, depois mais rapidamente; se o aumento da atividade enzimática muscular recidivar, a dose hormonal é aumentada novamente. Pacientes recuperados podem dispensar glicocorticoides, mas, na maioria das vezes, pacientes adultos requerem terapia com glicocorticoides a longo prazo (10 a 15 mg de prednisolona por dia). A dose inicial de prednisolona para crianças é de 30 a 60 mg/m² uma vez ao dia. Na presença de remissão por > 1 ano em crianças, a terapia com glicocorticoides pode ser descontinuada.
Em alguns casos, pacientes que recebem altas doses de glicocorticoides apresentam um aumento repentino na fraqueza muscular, o que pode estar associado ao desenvolvimento de miopatia glicocorticoide.
Em caso de resposta inadequada ao tratamento com glicocorticoides, bem como em caso de desenvolvimento de miopatia por glicocorticoides ou outras complicações que exijam redução da dose ou descontinuação da prednisolona, imunossupressores (metotrexato, ciclofosfamida, azatioprina, ciclosporina) devem ser utilizados. Alguns pacientes podem receber apenas metotrexato (geralmente em doses superiores às utilizadas no tratamento da AR) por mais de 5 anos. Imunoglobulinas intravenosas podem ser eficazes em pacientes refratários à terapia medicamentosa, mas seu uso aumenta o custo do tratamento.
Miosite associada a tumores primários e metastáticos, bem como miosite dos músculos do tronco, geralmente são mais refratárias à terapia com glicocorticoides. A remissão da miosite associada a tumores malignos é possível após a remoção do tumor.
Qual é o prognóstico para dermatomiosite e polimiosite?
Remissão a longo prazo (e até mesmo recuperação clínica) ao longo de 5 anos é observada em mais da metade dos pacientes tratados; em crianças, esse número é maior. A recidiva, no entanto, pode ocorrer a qualquer momento. A taxa de sobrevida global em cinco anos é de 75%, sendo maior em crianças. As causas de morte em adultos são fraqueza muscular grave e progressiva, disfagia, desnutrição, pneumonia por aspiração ou insuficiência respiratória devido a infecções pulmonares. A polimiosite é mais grave e resistente ao tratamento se houver danos ao coração e aos pulmões. A morte em crianças pode ocorrer devido a vasculite intestinal. O prognóstico geral da doença também é determinado pela presença de neoplasias malignas.