
Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
Síndrome de Tolosa-Hunt
Médico especialista do artigo

Última revisão: 04.07.2025
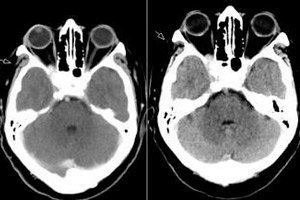
Síndrome da fissura orbital superior, oftalmoplegia patológica – tudo isso nada mais é do que a síndrome de Tolosa-Hunt, que é uma lesão das estruturas da fissura orbital superior. O processo geralmente envolve os vasos orbitais (arteriais e venosos), as fibras nervosas (nervos oculomotor, troclear, abducente, bem como o primeiro ramo do nervo trigêmeo) e o seio cavernoso adjacente. A doença pode ser classificada como uma patologia relativamente rara e bastante difícil de diagnosticar. [ 1 ]
Epidemiologia
A síndrome de Tolos-Hunt foi descrita há pouco tempo: cerca de 70 anos. Foi estudada pelo neurologista espanhol E. Tolos. Alguns anos depois, o trabalho foi complementado pelo oftalmologista inglês W. Hunt. Os nomes dos médicos-pesquisadores tornaram-se a base para o nome da síndrome.
A síndrome de Tolosa-Hunt é encontrada igualmente em homens e mulheres. A patologia geralmente é unilateral e observada com igual frequência no lado esquerdo ou direito. A síndrome bilateral é possível, mas ocorre apenas em casos isolados.
A idade média dos afetados é de 50 anos. Em geral, a síndrome de Tolosa-Hunt pode ser diagnosticada entre 15 e 85 anos. A maioria dos pacientes pertence à faixa etária idosa: o desenvolvimento da doença é facilitado por múltiplos distúrbios cardiovasculares, bem como por alterações teciduais relacionadas à idade.
O sintoma mais comum da doença é a manifestação de uma crise clássica de enxaqueca: a pessoa sente uma dor de cabeça súbita e pulsante em um dos lados, em "pontadas" ou "torções", com irradiação para a órbita ocular. Como a síndrome de Tolosa-Hunt não apresenta sintomas específicos típicos, a patologia é frequentemente chamada de "camaleão neurológico": o diagnóstico é complexo, exigindo diferenciação com muitas outras doenças.
Pacientes com a síndrome de Tolosa-Hunt são encontrados periodicamente em diferentes países do mundo, sem qualquer característica territorial ou sazonal. A taxa de incidência é de 0,3 a 1,5 casos por 1 milhão de habitantes. [ 2 ]
Causas Síndrome de Tolosa-Hunt
No decorrer da investigação das causas do desenvolvimento da síndrome de Tolosa Hunt, os cientistas descobriram os seguintes fatos:
- na maioria dos casos, a doença foi provocada por inflamação imunológica da parede externa do seio cavernoso;
- em alguns casos as causas foram malformações vasculares, processos tumorais no cérebro (formas primárias e secundárias), paquimeningite craniana localizada, miosite orbitária, periarterite nodosa e formação de trombos no seio cavernoso;
- Em aproximadamente 30% dos pacientes, a causa do distúrbio não pode ser determinada, então o diagnóstico de síndrome de Tolosa Hunt idiopática foi estabelecido.
Vamos considerar essas supostas razões com mais detalhes.
- O desenvolvimento autoimune da síndrome está associado tanto à hipotermia quanto a patologias infecciosas recentes, bem como a estresse profundo. A forma autoimune da doença é caracterizada por: início agudo, curso recorrente e alta eficácia da terapia com glicocorticosteroides. Esta forma da doença afeta mais frequentemente os homens.
- Malformações vasculares ocorrem frequentemente na hipertensão arterial descompensada. Mulheres são mais frequentemente afetadas. A doença tem início agudo, a dor é moderada e praticamente não há exoftalmia ou quemose.
- Dentre os processos tumorais capazes de levar ao desenvolvimento da síndrome de Tolosa-Hunt, os mais comuns foram tumores cerebrais primários, tumores metastáticos com focos primários nos pulmões, brônquios, próstata ou metástases de melanoma cutâneo.
- A paquimeningite craniana localizada causa um início agudo da síndrome na ausência de sinais cerebrais e meníngeos gerais, sem exoftalmia. O diagnóstico é confirmado morfologicamente durante biópsia.
- A miosite orbitária causa um início subagudo, com dor intensa e exoftalmia, quemose pronunciada e visão dupla.
- A trombose do seio cavernoso causa oftalmoplegia total. O diagnóstico é confirmado por ressonância magnética.
- A periarterite nodular pode causar o desenvolvimento da síndrome de Tolosa Hunt vários meses após o início da doença.
O mecanismo autoimune, na maioria dos casos, está subjacente à formação da patologia, o que foi comprovado por muitos especialistas. A natureza autoimune é indicada, em particular, pelos seguintes fatores:
- curso recorrente;
- transtornos dismúnicos;
- dissociação proteína-célula no líquido cefalorraquidiano e aumento dos níveis de citocinas pró-inflamatórias no líquido cefalorraquidiano e no soro sanguíneo. [ 3 ]
Fatores de risco
Os cientistas ainda não determinaram a causa exata da síndrome de Tolosa-Hunt. Mas conseguiram identificar alguns fatores que influenciam o desenvolvimento do transtorno:
- Predisposição genética a doenças autoimunes em geral. Se um dos membros da família sofreu ou sofre de uma doença autoimune, outros parentes também podem ter patologias semelhantes ou outras com um mecanismo de desenvolvimento semelhante. Este fator ainda é uma suposição que requer pesquisas e evidências adicionais.
- Fatores ambientais, incluindo hábitos alimentares, condições ambientais, qualidade da água, riscos industriais, etc.
- Situações estressantes severas, estresse frequente e choques psicoemocionais, alterações hormonais intensas (incluindo gravidez, menopausa, etc.).
- Doenças infecciosas crônicas de longo prazo, incluindo hepatite, infecção por herpesvírus, citomegalovírus, etc.
- Hipotermia, radiação, outros irritantes fortes e fatores prejudiciais.
Patogênese
O mecanismo etiológico do desenvolvimento da síndrome de Tolosa Hunt não foi totalmente esclarecido. O papel determinante é atribuído às reações autoimunes. Muitos cientistas presumem que infecções virais e microbianas, situações estressantes e radiação atuam apenas como fatores provocadores. Não há evidências sólidas da relação entre a entrada de microrganismos patogênicos no organismo e o desenvolvimento da síndrome de Tolosa Hunt. No entanto, há suspeitas sobre o envolvimento do citomegalovírus no processo autoimune, que contribui para a formação de granulomas. [ 4 ]
O esquema patogênico é causado pelo aparecimento de um processo inflamatório granulomatoso local na região da parede externa do seio cavernoso, porção infraclinoide ou supraclinoide da artéria carótida interna, que leva ao seu estreitamento. Um papel importante também é desempenhado pelo distúrbio da proteção imunológica humoral e celular. O lado humoral da síndrome está associado ao aumento da formação de anticorpos citoplasmáticos antineutrófilos que atuam contra as enzimas proteinase-3, mieloperoxidase e uma proteína de membrana específica capaz de se ligar a endotoxinas. Presumivelmente, os anticorpos citoplasmáticos estimulam os neutrófilos existentes, que, como resultado, atacam os órgãos "alvo"; em particular, o processo inflamatório se desenvolve na parede externa do seio cavernoso.
Alterações celulares também desempenham um papel no desenvolvimento da síndrome de Tolosa-Hunt, como demonstrado pela predominância de linfócitos T, macrófagos e plasmócitos nos granulomas.
Há informações sobre estruturas endoteliais altamente ativas e citocinas anti-inflamatórias, o que indica uma tendência do processo da doença se tornar crônico.
Em casos isolados, alterações necróticas focais foram observadas na área da parede externa do seio cavernoso.
Sintomas Síndrome de Tolosa-Hunt
Os sintomas característicos da síndrome de Tolosa-Hunt aparecem de forma repentina e inesperada para o paciente. Os principais sintomas são considerados os seguintes:
- Dor intensa na região da órbita ocular, extremamente desagradável, perfurante, que se espalha da região frontal para as sobrancelhas, para os olhos e para toda a cabeça.
- Visão dupla, que é detectada após o início da dor. Torna-se extremamente difícil para uma pessoa se concentrar visualmente e examinar qualquer objeto.
- O distúrbio da função motora do globo ocular, ou a chamada oftalmoplegia, é predominantemente unilateral. Pode se manifestar em graus variados, dependendo da gravidade do processo patológico e da extensão da lesão.
- Edema conjuntival.
- Deslocamento anterior do globo ocular (exoftalmia, olhos “esbugalhados”).
- Desvio do eixo visual de um globo ocular para o lado, estrabismo, típico de lesão nervosa unilateral.
- Deterioração geral da saúde, ligeiro aumento de temperatura, fraqueza, irritabilidade.
O quadro clínico progride gradualmente, os sintomas mudam e pioram, mas podem desaparecer tão repentinamente quanto surgiram. No entanto, na ausência da terapia necessária, a síndrome de Tolosa-Hunt volta a se manifestar com uma recaída.
Os sintomas neurológicos são causados pela localização do processo doloroso. A dor ocorre como resultado da irritação do primeiro ramo do nervo trigêmeo, que passa próximo ao tronco do nervo oculomotor, e é notada na região da órbita, testa, têmporas e base do nariz. A intensidade da dor varia de moderada a intensa.
Sintomas atípicos são possíveis, caracterizados pela ausência de dor. Isso pode ser observado quando a lesão está localizada antes da entrada do quinto par no seio cavernoso.
Os distúrbios oculomotores geralmente se manifestam como visão dupla durante o olhar direto.
Se o processo doloroso estiver localizado na região do ápice orbital, manifestações neurológicas são frequentemente encontradas em combinação com distúrbios do analisador visual. Como resultado, ocorre edema ou atrofia do disco do nervo óptico, e escotoma central é frequentemente observado. Exoftalmia (olhos esbugalhados) e quemose (edema conjuntival) são possíveis, cuja ocorrência é causada por alterações infiltrativas no tecido retrobulbar e dificuldades com o fluxo venoso da órbita.
Primeiros sinais
Como a síndrome de Tolosa-Hunt ainda não foi suficientemente estudada, os cientistas continuam a esclarecer os possíveis mecanismos de desenvolvimento desta patologia. Considerando os critérios delineados pela Sociedade Neurológica Internacional, o diagnóstico da síndrome de Tolosa-Hunt justifica-se na presença de um granuloma da parede externa do seio cavernoso, detetado durante ressonância magnética cerebral ou biópsia.
A lista de sinais que são aceitos como critérios diagnósticos para a síndrome é a seguinte:
- dor "em pontada" ou "torção" em uma órbita ocular com subsequente desenvolvimento de paralisia muscular (oftalmoplegia);
- lesões combinadas dos nervos oculomotores, do primeiro ramo do nervo trigêmeo e do plexo nervoso periarterial;
- um aumento do quadro clínico ao longo de vários dias (ou dentro de 1-2 semanas);
- a possibilidade de remissão espontânea (em alguns casos – com preservação residual dos defeitos);
- a probabilidade de uma recaída da síndrome, meses ou anos depois;
- quadro sistêmico inalterado, sem lesões fora do seio carotídeo;
- a presença de um efeito positivo da terapia com corticosteróides.
Há outra lista de características diagnósticas semelhantes proposta em 2003. De acordo com essa lista, a síndrome de Tolosa Hunt é considerada o resultado da proliferação de tecido granulomatoso no seio cavernoso, fissura orbital superior e cavidade orbital:
- um ou mais episódios de dor unilateral na área orbital que desaparecem sem tratamento por algumas semanas;
- dano ao nervo craniano (III, IV ou VI) na forma de paresia, presença de granuloma confirmada por ressonância magnética ou biópsia;
- o aparecimento de paresia simultaneamente com a síndrome dolorosa, ou dentro de 14 dias após ela;
- desaparecimento da paresia e da síndrome dolorosa dentro de 3 dias do início da terapia com corticoides.
Formulários
Na síndrome de Tolosa Hunt, os lados esquerdo e direito são afetados com frequência aproximadamente igual, por isso a patologia é dividida em lado esquerdo ou lado direito.
A doença geralmente é unilateral. Lesões bilaterais foram observadas apenas em casos extremamente raros.
O quadro clínico da doença pode evoluir através das seguintes fases:
- aguda ou subaguda, que ocorre após uma doença infecciosa viral recente, hipotermia, um forte aumento da pressão arterial, às vezes sem uma razão óbvia;
- recidiva crônica, com aumento gradual dos sintomas e exacerbações periódicas.
Além disso, a síndrome de Tolosa Hunt pode ser:
- total, com danos em todos os nervos que passam pela fissura orbitária superior;
- incompleto, com envolvimento no processo patológico dos nervos VI, IV, III pares e I ramo do V par em várias combinações.
Em relação ao seio, podem ser distinguidas as formas anterior, média e posterior da síndrome de Tolosa Hunt.
Complicações e consequências
A síndrome de Tolosa-Hunt é acompanhada por dor intensa, que acarreta perda de sono e distúrbios emocionais e mentais. Os doentes tornam-se irritáveis e emocionalmente instáveis. Se o tratamento necessário não for realizado, podem surgir distúrbios neuróticos nesse contexto: desenvolvem-se estados depressivos, neurastenia e hipocondria. A capacidade de trabalho é significativamente reduzida e o paciente torna-se retraído.
Uma característica da síndrome de Tolosa-Hunt é um curso recorrente, que frequentemente ocorre em doenças autoimunes. A duração do período de remissão pode ser muito variável: o indicador máximo registrado de duração assintomática foi de 11 anos. Após o tratamento, o risco de recidivas é significativamente reduzido. Se ocorrerem exacerbações, elas são menos graves.
Diagnósticos Síndrome de Tolosa-Hunt
Muitas vezes, torna-se difícil para os médicos diagnosticarem imediatamente a síndrome de Tolosa-Hunt, uma vez que os sintomas são muito semelhantes às manifestações de outras doenças mais comuns. Na maioria dos casos, é necessária uma consulta adicional com diversos especialistas: neurologista, oftalmologista, endocrinologista, oncologista, neurocirurgião, etc.
Na primeira fase, é necessário excluir doenças malignas, aneurismas, meningites, etc.
Na maioria das vezes, a síndrome de Tolosa-Hunt é diagnosticada por exclusão: o paciente é submetido a uma série de exames para descartar outras doenças mais prováveis. Os seguintes exames são necessários:
- hemograma detalhado;
- estudo da função hormonal da glândula tireoide;
- estudo do nível de proteína total no sangue (para avaliar a qualidade do metabolismo das proteínas);
- análise do líquido cefalorraquidiano.
- O diagnóstico instrumental envolve a realização dos seguintes procedimentos diagnósticos:
- ressonância magnética do cérebro e região orbital, com e sem contraste;
- angiografia por ressonância magnética;
- angiografia por subtração digital (angiografia por subtração intravenosa);
- Tomografia computadorizada de cérebro e órbita com e sem contraste.
A RM com realce de gadolínio é a modalidade de escolha para a avaliação da SCT e pode demonstrar aumento e realce anormais do EC, estendendo-se através da fissura orbital superior até o ápice orbital. Os achados de RM relatados em imagens ponderadas em T1 e T2 são extremamente variáveis e inespecíficos. A RM desempenha um papel fundamental no diagnóstico e ajuda a excluir outras lesões comuns associadas ao EC, evitando a necessidade de procedimentos invasivos de alto risco, como a biópsia de SC, a única maneira de obter confirmação histopatológica desta doença. [ 5 ]
Esses exames ajudam a identificar vestígios de processos inflamatórios no seio cavernoso, fissura orbital superior ou ápice orbital. Traços de inflamação na região orbital em imagens transversais na ausência de paralisia dos nervos cranianos são considerados mais benignos em termos de prognóstico.
Alguns pacientes com suspeita de síndrome de Tolosa Hunt são aconselhados a fazer uma biópsia para descartar câncer.
Diagnóstico diferencial
A prática clínica indica que sintomas semelhantes podem estar presentes em muitas patologias somáticas e neurológicas:
- em processos inflamatórios microbianos, virais e fúngicos que afetam as meninges ou a parede externa do seio cavernoso;
- em processos tumorais no cérebro e na órbita - por exemplo, em adenoma hipofisário, craniofaringioma, neurinoma, meningioma da asa do osso esfenoidal, em metástases cerebrais ou orbitais;
- em malformações vasculares - em particular, em aneurismas venoso-arteriais, fístulas carótido-cavernosas, etc., bem como em dissecções de ramos da artéria carótida interna;
- para trombose, formações císticas do seio cavernoso, linfoma;
- para sarcoidose, miosite orbitária (músculos oculares), granulomatose de Wegener (granulomatose com poliangeíte), oftalmomixarina e algumas patologias sanguíneas.
O diagnóstico diferencial envolve calcular a possibilidade de desenvolver todas as doenças acima, com base nos resultados de uma pesquisa, exame, estudos laboratoriais e instrumentais.
Na maioria das vezes, a síndrome de Tolosa Hunt deve ser diferenciada das seguintes patologias:
- bloqueio do seio cavernoso por um trombo;
- Síndrome de Rochon-Duvignod;
- síndrome do espaço retroesfenoidal (síndrome de Jacot);
- síndrome de Raeder paratrigeminal;
- polineuropatia craniana.
Quem contactar?
Tratamento Síndrome de Tolosa-Hunt
A síndrome de Tolosa-Hunt responde bem ao tratamento com um esquema imunossupressor de agentes hormonais corticosteroides. Esses medicamentos são capazes de suprimir a resposta agressiva do sistema imunológico e seus efeitos nocivos sobre os tecidos do corpo.
Os medicamentos mais comumente prescritos são Prednisolona, Metilprednisolona, Cortisona ou medicamentos alternativos que demonstraram efeitos positivos no tratamento de patologias autoimunes conhecidas. Os benefícios dos esteroides provavelmente estão relacionados ao mecanismo antioxidante e/ou à capacidade dessas altas doses de reduzir o edema e a isquemia subsequente nas áreas afetadas. [ 6 ]
Além dos corticosteroides, é apropriado o uso de analgésicos e anticonvulsivantes. Preparações multivitamínicas complexas são obrigatórias.
Se você seguir rigorosamente todas as instruções e recomendações do seu médico, os sintomas dolorosos da síndrome de Tolosa-Hunt são aliviados rapidamente: os pacientes notam uma melhora acentuada em seu bem-estar por volta do segundo ou terceiro dia. Na grande maioria dos casos, a capacidade de trabalho é mantida. [ 7 ]
As dosagens ideais e a frequência de uso de medicamentos hormonais são determinadas individualmente. Não existe um regime de tratamento universalmente aceito, visto que é muito difícil organizar estudos controlados por placebo, o que está associado à baixa prevalência da síndrome. Na maioria das vezes, altas doses de corticosteroides são recomendadas, embora tenham havido casos de eficácia com doses relativamente baixas de medicamentos (por exemplo, o uso de prednisolona em uma quantidade inferior a 0,5 mg/kg por dia). Atualmente, a dose média de prednisolona usada na síndrome de Tolosa-Hunt é de 1 a 2 mg/kg por dia.
Plano de tratamento aproximado:
- Metilprednisolona (Solu-Medron 1000 como infusão intravenosa gota a gota com 250 ml de solução isotônica de cloreto de sódio e Panangin (10,0) diariamente por cinco dias;
- Mildronato para normalização do metabolismo celular, 500 mg por injeção intravenosa em jato diariamente durante 10 dias;
- Neuromidin para melhorar a transmissão de impulsos ao longo das fibras neuromusculares, 20 mg por via oral três vezes ao dia;
- Clonazepam para aumentar o efeito inibitório na transmissão dos impulsos nervosos e estimulação dos receptores de benzodiazepínicos, 2 mg por via oral, e/ou Trileptal 150 mg por via oral antes de dormir.
É possível prescrever um curso prolongado de terapia com glicocorticosteroides usando altas doses de prednisolona. [ 8 ]
Prevenção
Não é possível prevenir a ocorrência da síndrome de Tolosa-Hunt antecipadamente. Isso se deve, pelo menos, ao fato de as causas da doença ainda não terem sido claramente determinadas. Se forem detectados quaisquer sintomas dolorosos – em particular, dor frequente na região frontal e nas órbitas oculares, visão dupla e enfraquecimento dos músculos oculares –, você deve entrar em contato com o especialista apropriado o mais rápido possível e realizar um diagnóstico completo.
A prevenção secundária visa prevenir recaídas em pacientes com Síndrome de Tolosa-Hunt já diagnosticada. Pontos importantes das ações preventivas são:
- consultas médicas regulares, procedimentos diagnósticos e acompanhamento ambulatorial especializado;
- cursos periódicos de terapia com corticosteróides;
- fortalecer e manter um estado adequado do sistema imunológico.
Todos aqueles que estão doentes devem tentar evitar situações estressantes e tratar prontamente qualquer processo inflamatório no corpo.
Previsão
O prognóstico da síndrome de Tolosa-Hunt é considerado favorável. Há uma boa resposta à corticoterapia, sendo comuns casos de remissão espontânea, embora alguns pacientes apresentem efeitos residuais na forma de comprometimento da função dos músculos oculares lesionados. Se não tratada, a doença torna-se subsequentemente recorrente. Em pacientes que receberam tratamento, recidivas são observadas em aproximadamente 35% dos casos. [ 9 ]
Após a conclusão do curso terapêutico, a capacidade de trabalho geralmente é restaurada. No entanto, isso se aplica a uma doença corretamente diagnosticada, e não a outras patologias que se desenvolvem sob a “máscara” da síndrome. [ 10 ]
A incapacidade é observada apenas em casos raros. Somente com exacerbações frequentes documentadas é possível atribuir o terceiro grupo de incapacidade. Em casos difíceis, o paciente é transferido para trabalhos leves, que não são acompanhados de estresse visual. Se a síndrome de Tolosa-Hunt for de natureza recorrente persistente, a pessoa não é recomendada a dirigir veículos, devido à função motora prejudicada dos globos oculares e à diplopia.