
Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
Xeroderma pigmentoso
Médico especialista do artigo

Última revisão: 04.07.2025
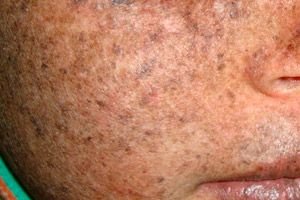
Xeroderma pigmentoso é uma doença genética autossômica recessiva que afeta o reparo do DNA. A doença é causada por mutações em genes envolvidos no reparo do DNA danificado pela radiação ultravioleta e outras substâncias tóxicas.
Na maioria das vezes, essa doença afeta crianças, também chamadas de "crianças da noite". Complicações frequentes da doença são carcinoma basocelular e outras neoplasias malignas da pele, melanoma maligno metastático e carcinoma espinocelular.
 [ 1 ]
[ 1 ]
Patogênese
A deficiência ou a ausência completa das enzimas endonucleases UV nas células do paciente (fibroblastos) desempenha um papel importante no desenvolvimento da doença. Essa enzima é responsável pela reprodução do DNA danificado pelos raios ultravioleta. De acordo com outras fontes, a deficiência da enzima DNA polimerase-1 desempenha um papel importante no desenvolvimento da doença em alguns pacientes. A doença se desenvolve mais frequentemente sob a influência de raios com comprimento de onda de 280-310 nm. Acredita-se que a xerodermia pigmentar se desenvolva devido à entrada de várias substâncias fotodinâmicas no corpo ou ao aumento de porfirinas no ambiente biológico humano.
Nos estágios iniciais da doença, observam-se hiperceratose, atrofia do foco epidérmico, afinamento da camada de Malpighi, aumento de grânulos de melanina na camada basal das células e infiltrado inflamatório crônico na camada superior da derme (principalmente ao redor dos vasos sanguíneos). Posteriormente, são detectadas alterações degenerativas nas fibras colágenas e elásticas e, no estágio tumoral, revelam-se sinais característicos de câncer de pele.
Sintomas xeroderma pigmentoso
Homens e mulheres são igualmente afetados por esta doença. A doença começa na primeira infância, na primavera ou no verão. A pele do paciente é especialmente sensível à luz solar. Portanto, a primeira erupção cutânea na forma de hiperpigmentação, semelhante a uma pinta, aparece nas áreas da pele expostas à luz solar. A erupção cutânea aumenta, o eritema intensifica-se, torna-se castanho-escuro, surgem telangiectasias, angiomas e atrofia. Posteriormente, crescem papilomas e verrugas (principalmente na pele da face e pescoço), e surgem manchas e úlceras. Como resultado da cicatrização da úlcera, o nariz torna-se mais fino ("bico de pássaro") e a pálpebra inverte-se. Os papilomas geralmente evoluem para tumores malignos. A xerodermia pigmentar, via de regra, ocorre em conjunto com o câncer espinocelular, os melanossarcomas.
O que está incomodando você?
Estágios
No curso clínico do xeroderma pigmentoso, distinguem-se cinco estágios: inflamatório (eritematoso), hiperpigmentação, atrofia, hiperceratose e tumores malignos.
Durante a fase inflamatória (eritematosa), surgem inchaço, manchas vermelhas e, às vezes, bolhas e vesículas nas áreas da pele expostas à luz solar (rosto, pescoço, parte superior do tórax, braços e mãos). Quase não há erupções cutâneas nas áreas não expostas à luz solar.
Na hiperpigmentação, manchas hiperpigmentadas marrom-claras, semelhantes a pintas, aparecem no lugar das manchas vermelhas.
Com a atrofia, a pele resseca, afina e enruga. Observam-se numerosas telangiectasias pequenas, estreladas, e cicatrizes com superfície brilhante na pele dos lábios e nariz. Devido à atrofia e às cicatrizes, desenvolvem-se microtomia (redução da abertura bucal), ectrópio, afinamento das orelhas e do nariz, atresia da abertura nasal e da boca. Em 80-85% dos pacientes, os olhos são afetados: observam-se conjuntivite, ceratite, danos à córnea e à mucosa e enfraquecimento da visão. Discromia, telangiectasia, alterações hiperceratóticas e tumores são observados na pele das pálpebras.
Com a hiperceratose, tumores verrucosos, papilomas, ceratoacantomas, fibromas, angiofibriomas e outros tumores benignos aparecem nos focos patológicos descritos acima. Por isso, alguns cientistas incluem a xerodermia pigmentar no grupo de doenças pré-cancerígenas.
Após 10 a 15 anos do início da doença, tumores malignos de pele (basalioma, endotelioma, angiossarcoma) surgem em focos atróficos alterados e manchas pigmentares. Os tumores são destruídos em pouco tempo, metastatizam para órgãos internos e levam à morte. Nos órgãos e tecidos internos de alguns pacientes, observam-se alterações distróficas gerais (sindactelia do segundo e terceiro dedos, distrofia dentária, perda completa de pelos, etc.).
Formulários
A forma neurológica do xeroderma pigmentoso se manifesta em duas síndromes.
A síndrome de Reed é caracterizada por manifestações clínicas de xeroderma pigmentoso e microcefalia, idiopatia e crescimento lento do sistema esquelético. Na forma neurológica da doença, a reparação do DNA é difícil e a radioterapia leva ao aumento dos principais focos patológicos.
A síndrome De Sinctis-X Cocchione é caracterizada pelos seguintes sinais clínicos:
- desenvolvimento de sinais clínicos de xeroderma pigmentoso em pele particularmente fotossensível;
- aparecimento precoce de tumores malignos;
- curso simultâneo de paralisia espástica, microcefalia e demência;
- deformidades congênitas e nanismo;
- subdesenvolvimento das gônadas;
- abortos frequentes;
- transmissão recessiva por herança.
Segundo alguns dermatologistas, a síndrome de Santis-Cacchione não é uma doença independente, mas uma manifestação clínica grave e totalmente expressa do xeroderma pigmentoso.
Formas genéticas
Tipos |
Gene |
Locus |
Descrição |
Tipo A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentoso (XP) grupo A – forma clássica |
Tipo B, II, XPB |
XPB |
2º trimestre de 2021 |
XP Grupo B |
Tipo C, III, XPC |
XPC |
3p25 |
XP Grupo C |
Tipo D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP Grupo D ou Síndrome de De Sanctis-Cacchione |
Tipo E, V, XPE |
DDB2 |
11p12-p11 |
XP Grupo E |
Tipo F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
XP Grupo F |
Tipo G, VII, XPG |
RAD2ERCC5 |
13q33 |
Grupo XP G e síndrome COFS (síndrome cérebro-óculo-facioesquelética) tipo 3 |
Tipo V, XPV |
POLH |
6p21.1-p12 |
Xeroderma Pigmentosa Variante |
O que precisa examinar?
Como examinar?
Quem contactar?
Tratamento xeroderma pigmentoso
Medicamentos antimaláricos (delagyl, plaquenil, resoquin, etc.), protegendo o DNA dos raios ultravioleta, inibem o processo de despolimerização e reduzem a sensibilidade (fotossensibilização) da pele à luz solar. Esses medicamentos possuem propriedades anti-inflamatórias e hipossensibilizantes. Recomenda-se a realização de terapia geral em conjunto com terapia vitamínica (B1, B2, PP, B6, B12, A, E), anti-histamínicos (tavegil, difenidramina, suprastina) e agentes dessensibilizantes (tiossulfato de sódio, cloreto de cálcio a 10%, 10 ml por via intravenosa).
Para tratamento local, são utilizados cremes e pomadas com proteção solar.
Na forma tumoral da xerodermia pigmentar, são utilizados métodos cirúrgicos, nitrogênio líquido e raios laser. Para se proteger da luz solar, é necessário usar roupas largas, chapéus e luvas.
Previsão
A maioria dos pacientes (2/3) morre antes de completar 15 anos. Alguns pacientes podem viver até 40-50 anos.
[ 20 ]