
Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
Encefalomiopatia necrosante subaguda de Leah
Médico especialista do artigo

Última revisão: 04.07.2025
A doença foi mencionada pela primeira vez em 1951. Até o momento, mais de 120 casos foram descritos. A doença de Leigh (OMIM 256000) é uma doença geneticamente heterogênea que pode ser herdada nuclearmente (autossômica recessiva ou ligada ao cromossomo X) ou mitocondrialmente (menos comum).
 [
[ Causas da síndrome de Leah
A doença se baseia na deficiência de enzimas responsáveis pela produção de energia, principalmente devido à interrupção do metabolismo do ácido pirúvico e a um defeito no transporte de elétrons na cadeia respiratória. Desenvolve-se uma deficiência do complexo piruvato desidrogenase (subunidade a-E1), piruvato carboxilase, complexo 1 (NAD-coenzima Q-redutase) e complexo 4 (citocromo oxidase) da cadeia respiratória.
Foi estabelecido que defeitos da piruvato carboxilase, complexo 1 (NAD-coenzima Q-redutase) e complexo 4 (citocromo oxidase) da cadeia respiratória são herdados de forma autossômica recessiva, defeitos do complexo piruvato desidrogenase (subunidade a-E1) são herdados de forma recessiva ligada ao cromossomo X. No caso de mutações pontuais do mtDNA, que afetam a 6ª subunidade da ATPase, a herança mitocondrial é típica. Na maioria das vezes, ocorre uma mutação miscens, associada à substituição da timina por guanina ou citosina na posição 8993 do mtDNA. Menos comum é uma mutação na posição 9176 do mtDNA. Devido ao fato de a mutação T8993G ser o principal defeito na síndrome NARP, famílias com essas duas doenças foram descritas. Em crianças, também foi descrita uma mutação no mtDNA na posição 8344, que ocorre na síndrome MERRF.
Supõe-se que, no caso de acúmulo de mtDNA mutante na maioria das mitocôndrias, se desenvolva um curso grave da síndrome de Leigh. Na gênese mitocondrial dessa condição, o mtDNA mutante é encontrado em 90% de todas as mitocôndrias. A patogênese está associada à violação da formação de energia nas células e ao desenvolvimento de acidose láctica.
Sintomas da síndrome de Leah
Os primeiros sinais da doença surgem em idade precoce (1 a 3 anos). No entanto, são conhecidos casos de manifestação da doença às 2 semanas e aos 6 a 7 anos de idade. Inicialmente, desenvolvem-se distúrbios inespecíficos: atraso no desenvolvimento psicomotor, diminuição do apetite, episódios de vômitos e déficit de peso corporal. Posteriormente, aumentam os sintomas neurológicos: hipotonia ou distonia muscular com transição para hipertonia, crises de mioclonia ou convulsões tônico-clônicas, tremor das extremidades, coreoatetose, distúrbio de coordenação, diminuição dos reflexos tendinosos, letargia e sonolência. A neurodegeneração cerebral é progressiva. Os sintomas de insuficiência piramidal e extrapiramidal aumentam e a deglutição é prejudicada. Alterações no órgão da visão, como ptose palpebral, oftalmoplegia, atrofia do nervo óptico e, menos frequentemente, degeneração pigmentar da retina, são frequentemente observadas. Às vezes, desenvolve-se cardiomiopatia hipertrófica e surgem episódios de taquipneia.
Raramente, a doença evolui como uma encefalopatia aguda. Mais comumente, a evolução é crônica ou subaguda, levando à morte vários anos após o início da doença. Com uma evolução rápida (várias semanas), a morte ocorre como resultado da paralisia do centro respiratório.
Diagnósticos da síndrome de Leah
Um exame bioquímico de sangue revela acidose láctica devido ao acúmulo de ácidos láctico e pirúvico no sangue e no líquido cefalorraquidiano, bem como um aumento no teor de alanina no sangue. Os níveis de corpos cetônicos também podem estar elevados. A urina detecta aumento da excreção de ácidos orgânicos: láctico, fumárico, etc. Os níveis de carnitina no sangue e nos tecidos frequentemente diminuem.
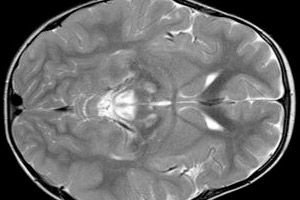
Os resultados do EEG revelam sinais focais de atividade epiléptica. Os dados da ressonância magnética revelam dilatação dos ventrículos cerebrais, lesão cerebral bilateral e calcificação dos gânglios da base (núcleo caudado, putâmen, substância negra e globo pálido). Atrofia dos hemisférios cerebrais e da substância encefálica também pode ser detectada.
O exame morfológico revela alterações macroscópicas na substância cerebral: focos simétricos de necrose, desmielinização e degeneração esponjosa do cérebro, principalmente das seções médias, ponte, gânglios da base, tálamo e nervo óptico. O quadro histológico inclui degeneração cística do tecido cerebral, gliose astrocítica, morte neuronal e aumento do número de mitocôndrias nas células. Nos músculos esqueléticos, há acúmulo de inclusões lipídicas, diminuição da reação histoquímica aos complexos 1 e 4 da cadeia respiratória, acúmulo subsarcolemal de mitocôndrias, mitocôndrias anormais com desorganização das cristas. O fenômeno da FRR frequentemente não é detectado.
Como examinar?
Quais testes são necessários?