
Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
Novas descobertas contribuem para uma melhor compreensão das causas da síndrome de Rett
Última revisão: 02.07.2025
 ">
">A síndrome de Rett é um distúrbio raro do neurodesenvolvimento para o qual atualmente não há cura ou tratamento adequado. Ela causa sintomas físicos e cognitivos graves, muitos dos quais se sobrepõem aos transtornos do espectro autista.
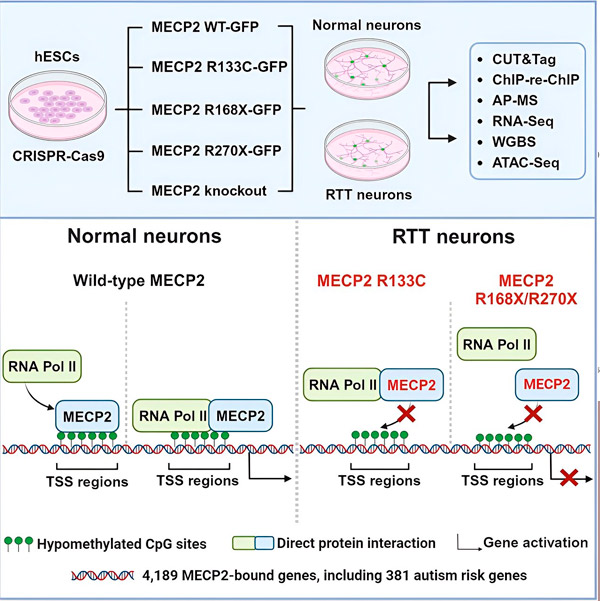
A síndrome de Rett é causada por mutações no gene MECP2, altamente expresso no cérebro e que parece desempenhar um papel importante na manutenção da saúde dos neurônios. O gene está localizado no cromossomo X e a síndrome afeta principalmente meninas. Para desenvolver tratamentos para a síndrome de Rett, os pesquisadores querem entender melhor o MECP2 e suas funções no cérebro.
Pesquisadores, incluindo o cofundador do Instituto Whitehead, Rudolf Jaenisch, estudam o MECP2 há décadas, mas muitos fatos básicos sobre o gene permanecem desconhecidos. A proteína codificada pelo gene, MECP2, está envolvida na regulação gênica; ela se liga ao DNA e influencia os níveis de expressão de vários outros genes, ou a quantidade de proteína que eles produzem.
No entanto, os pesquisadores não tinham uma lista completa de genes afetados pelo MECP2, e não havia consenso sobre como o MECP2 afeta esses genes.
Estudos iniciais sobre a MECP2 sugeriram que ela atuava como um repressor, reduzindo a expressão de seus genes-alvo, mas pesquisas de Jaenisch e outros já haviam demonstrado que a MECP2 também atua como um ativador, aumentando a expressão de seus alvos — e que pode ser um ativador em primeiro lugar. Também não se sabia o mecanismo de ação da MECP2, ou o que exatamente a proteína faz para causar alterações na expressão gênica.
Limitações tecnológicas impediram que pesquisadores esclarecessem essas questões. Mas Yanish, o pós-doutorado de seu laboratório, Yi Liu, e o ex-membro do laboratório de Yanish, Anthony Flamier, agora professor assistente no centro de pesquisa CHU Sainte-Justine da Universidade de Montreal, usaram técnicas de ponta para responder a essas questões remanescentes sobre o MECP2 e obter novos insights sobre seu papel na saúde e nas doenças cerebrais.
Seus resultados foram publicados no periódico Neuron e os pesquisadores também criaram um repositório on-line de seus dados MECP2, o portal MECP2-NeuroAtlas, como um recurso para outros pesquisadores.
"Acredito que este artigo mudará fundamentalmente a compreensão das pessoas sobre como o MECP2 causa a síndrome de Rett. Temos uma compreensão completamente nova do mecanismo, e isso pode abrir novos caminhos para o desenvolvimento de tratamentos para a doença", afirma Janisch, que também é professor de biologia no MIT.
Compreensão mais profunda do MECP2 no cérebro
Os pesquisadores primeiro criaram um mapa detalhado de onde o MECP2 se liga em sequências de genes neuronais humanos, seja dentro de genes ou em regiões regulatórias de DNA próximas a eles. Eles usaram uma abordagem chamada CUT&Tag, que pode identificar interações de proteínas com o DNA com alta precisão.
Os pesquisadores encontraram mais de 4.000 genes associados ao MECP2. Eles repetiram o mapeamento em neurônios com mutações comuns do MECP2 associadas à síndrome de Rett para determinar onde o MECP2 está esgotado no estado da doença.
Saber a quais genes o MECP2 se liga permitiu que Liu e Flamier começassem a estabelecer conexões entre os alvos do MECP2 e a saúde cerebral. Eles descobriram que muitos de seus alvos estão envolvidos no desenvolvimento e na função de axônios e sinapses neuronais.
Eles também compararam sua lista de alvos do MECP2 com o banco de dados de genes associados ao autismo da Simons Foundation Autism Research Initiative (SFARI) e descobriram que 381 genes naquele banco de dados são alvos do MECP2.
Fonte: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Essas descobertas podem ajudar a esclarecer os mecanismos subjacentes aos sintomas do autismo na síndrome de Rett e fornecer um bom ponto de partida para investigar o possível papel do MECP2 no autismo.
"Criamos o primeiro mapa integrado do epigenoma MECP2 na saúde e na doença, e este mapa pode orientar pesquisas futuras", afirma Liu. "Saber quais genes são alvos do MECP2 e quais genes são diretamente afetados na doença fornece uma base sólida para a compreensão da síndrome de Rett e para questionar a regulação gênica em neurônios."
Os pesquisadores também analisaram se o MECP2 aumentava ou diminuía a expressão de seus genes-alvo. Em consonância com o histórico de MECP2 ser identificado por alguns como ativador e por outros como repressor, Liu e Flamier encontraram exemplos em que o MECP2 desempenhava ambas as funções.
No entanto, embora o MECP2 seja mais frequentemente considerado um repressor, Liu e Flamier descobriram que ele atua principalmente como um ativador — confirmando descobertas anteriores de Jaenisch e Liu. Um novo experimento mostrou que o MECP2 ativa pelo menos 80% de seus alvos, e outro descobriu que ele ativa até 88% de seus alvos.
O mapa de genes-alvo criado pelos pesquisadores forneceu informações adicionais sobre o papel do MECP2 como ativador. Eles descobriram que, para os genes que o MECP2 ativa, ele normalmente se liga a uma região do DNA a montante do gene, chamada de sítio de início da transcrição.
Este é o local onde a maquinaria celular inicia o processo de transcrição de um gene em RNA, após o qual o RNA é traduzido em uma proteína funcional, que é o produto da expressão gênica. A presença de MECP2 no local de início da transcrição, onde a expressão gênica se inicia, é consistente com seu papel como ativador gênico.
Os pesquisadores então se propuseram a determinar o papel da MECP2 na ativação gênica. Eles analisaram as moléculas às quais a MECP2 se liga neste sítio, além do DNA, e descobriram que a MECP2 interage diretamente com um complexo proteico chamado RNA polimerase II (RNA Pol II). A RNA Pol II é uma máquina celular essencial que transcreve DNA em RNA. A RNA Pol II não consegue encontrar genes por conta própria, por isso requer uma variedade de cofatores, ou proteínas colaboradoras, para ajudá-la a realizar seu trabalho.
Os pesquisadores propõem que o MECP2 atue como um desses cofatores, auxiliando o RNA Pol II a iniciar a transcrição em genes onde o MECP2 se liga. A análise estrutural do MECP2 identificou partes da molécula que se ligam ao RNA Pol II, e outros experimentos confirmaram que a perda do MECP2 reduz a presença do RNA Pol II em locais apropriados para o início da transcrição, bem como os níveis de expressão dos genes-alvo.
Isso sugere que a síndrome de Rett pode ser causada pela diminuição da transcrição de genes alvos da MECP2 devido a mutações na MECP2 que a impedem de se ligar à RNA Pol II ou ao DNA. Em consonância com essa ideia, as mutações mais comuns na MECP2 associadas à doença são truncamentos: mutações nas quais parte da proteína está ausente, o que pode alterar a interação entre a MECP2 e a RNA Pol II.
Os pesquisadores esperam que suas descobertas não apenas mudem nossa compreensão do MECP2, mas que uma compreensão mais profunda e ampla de como o MECP2 influencia o desenvolvimento e a função cerebral possa levar a novos insights que ajudarão pessoas com síndrome de Rett e distúrbios relacionados, incluindo autismo.
"Este projeto é um ótimo exemplo da natureza colaborativa do laboratório Janisch", diz Flamier. "Rudolf e eu tínhamos um problema específico relacionado à síndrome de Rett, e eu tinha experiência com a tecnologia CUT&Tag, que poderia resolvê-lo. Através da discussão, percebemos que poderíamos unir nossos esforços e agora temos um ótimo repositório de informações sobre o MECP2 e suas ligações com a doença."