
Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
A alcaptonúria é uma anomalia enzimática congénita
Médico especialista do artigo

Última revisão: 04.07.2025
Um dos distúrbios metabólicos muito raros, a alcaptonúria, refere-se a anomalias congênitas no metabolismo do aminoácido tirosina.
Esta síndrome também pode ser chamada de deficiência de homogentisato oxidase, homogentisinúria, ocronose hereditária ou doença da urina preta.[ 1 ]
Epidemiologia
Segundo as estatísticas, não há mais de nove casos de alcaptonúria por milhão de pessoas. E na maioria dos países europeus, há um caso para cada 100 a 250 mil nascidos vivos.
Entre os países europeus, a exceção é a Eslováquia (especialmente a relativamente pequena região noroeste), onde a prevalência de alcaptonúria é de um caso a cada 19.000 recém-nascidos. Isso provavelmente se deve ao fato de que, entre as famílias ciganas eslovacas que vivem lá, o nível de endogamia (casamento entre primos) é o mais alto da Europa: 10-14%. [ 2 ]
Causas alcaptonúria
As causas exatas da alcaptonúria, como um distúrbio congênito do catabolismo (degradação metabólica) do α-aminoácido aromático (homocíclico) tirosina, foram estabelecidas: esse tipo de distúrbio metabólico é consequência de mutações homozigotas ou heterozigotas compostas de um dos milhares de genes no cromossomo 3, mais precisamente, o gene HGD no locus 3q21-q23 no braço longo do cromossomo. Este gene codifica as sequências de nucleotídeos da enzima hepática homogentisato-1,2-dioxigenase [ 3 ] (também chamada de oxidase do ácido homogentísico ou oxidase homogentisato) - uma metaloproteína contendo ferro necessária para um dos estágios da degradação da tirosina no corpo. [ 4 ], [ 5 ]
Assim, a alcaptonúria é um defeito da enzima homogentisato-1,2-dioxigenase, ou mais precisamente, o resultado de sua deficiência geneticamente determinada ou ausência completa. [ 6 ]
Por ser uma deficiência enzimática congênita, a alcaptonúria é herdada como um traço autossômico recessivo, ou seja, para que a alcaptonúria ocorra em crianças, ambos os pais devem ter um gene modificado para a enzima, já que cada um deles passa para o filho apenas uma cópia do gene das duas disponíveis.
De acordo com os dados mais recentes, existem mais de duzentas variantes de modificação do gene HGD, sendo as mutações sem sentido, a translocação e o splicing as mais frequentemente observadas.
Fatores de risco
O único fator de risco para o desenvolvimento desta enzimopatia congênita é a sua presença na história familiar e a herança de duas cópias modificadas do gene HGD, caso os pais não apresentem alcaptonúria (o risco de transmissão da anomalia é de 25%), ou um dos pais tenha esta doença. [ 7 ]
Patogênese
A tirosina desempenha um papel fundamental na síntese de proteínas, na produção de cromoproteínas – o pigmento da pele melanina – bem como nos hormônios tireoidianos e nos neurotransmissores catecolaminas.
O mecanismo de regulação da quantidade de tirosina nas células é muito complexo, e o corpo normaliza seu excesso de conteúdo decompondo-o. O processo de catabolismo da tirosina, como o de todos os aminoácidos aromáticos, é multifásico e ocorre em várias etapas. Cada etapa da degradação metabólica da tirosina ocorre com a participação de uma enzima específica e a formação de um composto intermediário.
Assim, primeiro o aminoácido é decomposto em para-hidroxifenilpiruvato, que é convertido em alcaptona – ácido 2,5-di-hidroxifenilacético ou homogentísico. Em seguida, a alcaptona deveria ser transformada em ácido maleacético, mas isso não ocorre. [ 8 ]
E a patogênese da alcaptonúria consiste na cessação das reações bioquímicas do catabolismo da tirosina na fase de formação do ácido homogentísico: simplesmente não há enzima necessária para quebrá-lo – a homogentisato oxidase.
O ácido homogentísico não é utilizado pelo organismo e pode acumular-se com a excreção renal. Além disso, é oxidado em benzoquinoacetato (ácido benzoquinonacético), que, ao se ligar às moléculas dos tecidos e fluidos corporais, forma compostos biopolímeros com coloração semelhante à melanina.
O acúmulo desses produtos intermediários no tecido leva a uma ruptura da estrutura de colágeno do tecido cartilaginoso, o que reduz sua elasticidade – com o aparecimento de muitos sinais clínicos de alcaptonúria e o desenvolvimento de complicações.
Sintomas alcaptonúria
A alcaptonúria em recém-nascidos e lactentes é caracterizada pelo escurecimento da urina. Quando exposta ao ar, a urina em fraldas, fraldas e roupas íntimas torna-se marrom-escura; isso se deve ao acúmulo e à liberação de ácido homogentísico, que é oxidado a benzoquinoacetato. [ 9 ]
Na ausência de outros sintomas, a alcaptonúria em crianças pequenas frequentemente não é diagnosticada a tempo, pois a urina pode escurecer após várias horas de micção. Segundo alguns dados, apenas um quinto das crianças menores de 12 meses que nascem com deficiência dessa enzima são identificadas em ambientes clínicos. Portanto, é muito importante que os pais prestem atenção aos cuidados com seus bebês.
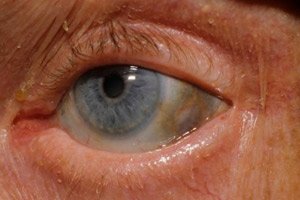
Além disso, os primeiros sinais incluem pigmentação (cor cinza-azulada) da esclera dos olhos e das cartilagens das orelhas e do nariz, o que geralmente é chamado de ocronose.[ 10 ]
Com o tempo, outros sintomas aparecem:
- pigmentação severa da pele nas maçãs do rosto, axilas e genitais;
- manchas nas roupas quando em contato com áreas suadas do corpo;
- ataques de fraqueza geral;
- voz rouca.
Deve-se ter em mente que alcaptonúria e ocronose, como observado acima, são nomes sinônimos para o mesmo distúrbio do catabolismo da tirosina.
Doença da urina em xarope de bordo e alcaptonúria. A doença da urina em xarope de bordo congênita, ou leucinose, também é um distúrbio metabólico, tem o mesmo padrão de herança e até mesmo mutações ocorrem no mesmo cromossomo, mas afetam o gene que codifica o complexo enzimático da α-cetoácido desidrogenase de cadeia ramificada. Por causa disso, o corpo não consegue decompor certos componentes das proteínas, em particular, os aminoácidos leucina, isoleucina e valina. Com essa doença, a urina (e a cera de ouvido) têm um odor adocicado; além disso, o quadro clínico desse tipo de acidemia orgânica inclui hipopigmentação, flutuações na pressão arterial, convulsões, vômitos e diarreia, queda nos níveis de glicose no sangue, cetoacidose, alucinações, etc. A taxa de mortalidade em crianças é bastante alta; em adultos, sem tratamento, coma e morte podem ocorrer devido a edema cerebral.
Albinismo e alcaptonúria são "unidos" apenas pela tirosina. O albinismo, incluindo o oculocutâneo, é causado por mutações genéticas que afetam a produção do pigmento melanina. Alterações congênitas são observadas no gene TYR no cromossomo 11 (11q14.3), que codifica a tirosinase, uma enzima melanossomal contendo cobre, necessária para a formação do pigmento da pele com base nos produtos do metabolismo da tirosina. Esta doença é muito mais comum do que a alcaptonúria.
Complicações e consequências
Causadas pela ação de metabólitos intermediários da tirosina – ácidos homogentísico e benzoquinoneacético – as consequências e complicações da alcaptonúria surgem devido à deposição de polímeros pigmentados reativos, à destruição de fibrilas de colágeno e à deterioração do estado da cartilagem (com diminuição de sua resistência ao estresse mecânico).
Com o passar dos anos, na idade adulta, desenvolvem-se artrite degenerativa e osteoartrite de grandes articulações (quadril, sacroilíaca e joelho); os espaços intervertebrais estreitam-se (especialmente na coluna lombar e torácica) – com calcificação e formação de osteófitos; a densidade do tecido das placas ósseas subcondrais diminui e os ossos subjacentes podem sofrer remodelação patológica com a formação de crescimentos e deformações. [ 11 ]
Podem ser observados danos nas válvulas cardíacas (aórtica e mitral) e nas artérias coronárias – com sinais de doença arterial coronariana, bem como formação de cálculos nos rins e na próstata – devido à mesma calcificação. [ 12 ], [ 13 ]
Diagnósticos alcaptonúria
Normalmente, o diagnóstico de distúrbios metabólicos congênitos é baseado no estudo de fluidos biológicos do corpo.
Com base em quais testes e reações a alcaptonúria pode ser diagnosticada? Exames de urina são necessários para detectar o ácido homogentísico e determinar seu nível (normal – 20-30 mg por dia, elevado – 3-8 g). Uma amostra de urina é examinada por cromatografia gasosa ou espectrometria de massa, utilizando cromatografia líquida; um teste de triagem para a presença de cloreto de ferro na urina é possível. [ 14 ]
Existe também um método para diagnóstico rápido – determinação de alcapton em manchas de urina seca em papel (pela intensidade da cor).
Para esclarecer o diagnóstico, o diagnóstico instrumental (radiografia) envolve a identificação de sinais radiológicos de osteoartrite e outras patologias articulares em pacientes.
O diagnóstico é confirmado por métodos genéticos moleculares de diagnóstico de doenças hereditárias, como testes genéticos e sequenciamento de DNA. [ 15 ]
Diagnóstico diferencial
O diagnóstico diferencial inclui hemocromatose e insuficiência hepática aguda do recém-nascido, melaninúria, porfiria aguda intermitente, linfo-histiocitose hemofagocítica, patologia mitocondrial primária, artrite reumatoide, espondilite anquilosante.
Quem contactar?
Tratamento alcaptonúria
O principal tratamento para a alcaptonúria é a administração oral de altas doses (pelo menos 1000 mg por dia) de ácido ascórbico. Em crianças, isso aumenta a excreção de ácido homogentísico na urina e, em adultos, reduz o conteúdo urinário de seu derivado, o ácido benzoquinonacético, e retarda sua ligação às estruturas do tecido conjuntivo das articulações e ao colágeno. [ 16 ]
Clínicas da Europa Ocidental estão testando o medicamento Nitisinona (Orfalin), um medicamento do grupo de metabólitos que inibe a segunda etapa do catabolismo da tirosina: a transformação do para-hidroxifenilpiruvato em ácido homogentísico. No entanto, o uso desse agente farmacológico leva ao acúmulo de tirosina e pode causar efeitos colaterais graves, incluindo opacidade da córnea e fotofobia, sangramento nasal e estomacal, insuficiência hepática, alterações no sangue, etc. No entanto, nos Estados Unidos, a Nitisinona é aprovada pela FDA para o tratamento da tirosinemia tipo I. [17 ], [ 18 ]
Assim, o tratamento fisioterapêutico – terapia por exercícios para aumentar a força muscular e melhorar a mobilidade articular, balneoterapia e terapia peloide para limitar a dor – é realizado para problemas articulares causados pela alcaptonúria.
Embora a tirosina não seja fornecida apenas pelos alimentos, mas também produzida pelo corpo, recomenda-se que pacientes com alcaptonúria sigam uma dieta com baixo teor de proteína e limitem o consumo de alimentos ricos em tirosina, principalmente carne bovina e suína, laticínios (especialmente queijos), legumes, nozes e sementes.
Prevenção
A prevenção de mutações genéticas é impossível, mas para evitar o nascimento de crianças com alto risco de doenças congênitas, existe o aconselhamento genético médico, necessário antes de uma gravidez planejada para aqueles casais cujo histórico familiar inclui doenças hereditárias. [ 19 ]
Previsão
Resultados fatais da alcaptonúria são muito raros, e a morte pode ser causada por complicações graves envolvendo o coração e os rins. Portanto, a expectativa de vida geral de pessoas com alcaptonúria é boa.
Mas a qualidade de vida é reduzida devido a dores intensas nas articulações ou na coluna com limitação significativa da mobilidade, muitas vezes progressiva.