
Todo o conteúdo do iLive é medicamente revisado ou verificado pelos fatos para garantir o máximo de precisão factual possível.
Temos diretrizes rigorosas de fornecimento e vinculamos apenas sites de mídia respeitáveis, instituições de pesquisa acadêmica e, sempre que possível, estudos médicos revisados por pares. Observe que os números entre parênteses ([1], [2], etc.) são links clicáveis para esses estudos.
Se você achar que algum dos nossos conteúdos é impreciso, desatualizado ou questionável, selecione-o e pressione Ctrl + Enter.
Fibrose cística
Médico especialista do artigo

Última revisão: 04.07.2025
A fibrose cística é uma doença genética monogênica autossômica recessiva caracterizada por um distúrbio da secreção de glândulas exócrinas de órgãos vitais com danos principalmente aos sistemas respiratório e digestivo, curso grave e prognóstico desfavorável.
 [ 1 ]
[ 1 ]
Epidemiologia
A incidência de fibrose cística oscila entre 1:2.500 e 1:4.600 recém-nascidos. Anualmente, nascem cerca de 45.000 pessoas com fibrose cística em todo o mundo. A incidência de portadores do gene da fibrose cística é de 3% a 4%, com cerca de 275 milhões de pessoas em todo o mundo sendo portadoras desse gene, das quais cerca de 5 milhões vivem na Rússia e cerca de 12,5 milhões nos países da CEI.
Causas fibrose cística
A fibrose cística é transmitida de forma autossômica recessiva. O gene da fibrose cística está localizado no autossomo 7, contém 27 éxons e consiste em 250.000 pares de nucleotídeos.
Um único gene pode apresentar muitas mutações, cada uma delas específica para uma determinada população ou região geográfica. Mais de 520 mutações foram descritas, sendo a mais comum a delta-P-508, ou seja, uma substituição do aminoácido fenilalanina na posição 508.
Patogênese
Mutações no gene da fibrose cística alteram a estrutura e a função de uma proteína chamada CFTR (regulador transmembrana da fibrose cística). Essa proteína atua como um canal de cloreto envolvido na troca hidroeletrolítica das células epiteliais do sistema broncopulmonar, trato gastrointestinal, pâncreas, fígado e sistema reprodutor. Como resultado da alteração da função e da estrutura da proteína CFTR, íons cloreto Cl- acumulam -se no interior da célula. Isso leva a uma alteração no potencial elétrico no lúmen dos ductos excretores, o que facilita o fluxo de grandes quantidades de íons sódio (Na + ) do lúmen do ducto para a célula e aumenta ainda mais a absorção de água do espaço pericelular.
Como resultado dessas alterações, a secreção da maioria das glândulas exócrinas engrossa, sua evacuação é interrompida, o que leva a distúrbios secundários pronunciados em órgãos e sistemas, mais pronunciados nos sistemas broncopulmonar e digestivo.
Um processo inflamatório crônico de intensidade variável se desenvolve nos brônquios, a função do epitélio ciliado é gravemente prejudicada, o escarro torna-se muito viscoso, espesso e muito difícil de evacuar, sua estagnação é observada, bronquiolite e bronquiectasias se formam, as quais se tornam mais comuns com o tempo. Essas alterações levam ao aumento da hipóxia e à formação de cardiopatia pulmonar crônica.
Pacientes com fibrose cística são extremamente predispostos ao desenvolvimento de inflamação crônica no sistema broncopulmonar. Isso se deve a distúrbios pronunciados no sistema de defesa broncopulmonar local (níveis reduzidos de IgA, interferon, função fagocitária de macrófagos alveolares e leucócitos).
Os macrófagos alveolares desempenham um papel importante no desenvolvimento da inflamação crônica no sistema broncopulmonar. Eles produzem grandes quantidades de IL-8, o que aumenta drasticamente a quimiotaxia dos neutrófilos na árvore brônquica. Os neutrófilos acumulam-se em grandes quantidades nos brônquios e, juntamente com as células epiteliais, secretam muitas citocinas pró-inflamatórias, incluindo IL-1, IL-8, IL-6, fator de necrose tumoral e leucotrienos.
A alta atividade da enzima elastase também desempenha um papel importante na patogênese dos danos ao sistema broncopulmonar. Distingue-se entre elastase exógena e endógena. A primeira é produzida pela flora bacteriana (especialmente Pseudomonas aeruginosa), a segunda, por leucócitos neutrófilos. A elastase destrói o epitélio e outros elementos estruturais dos brônquios, o que contribui para uma maior interrupção do transporte mucociliar e para a rápida formação de bronquiectasias.
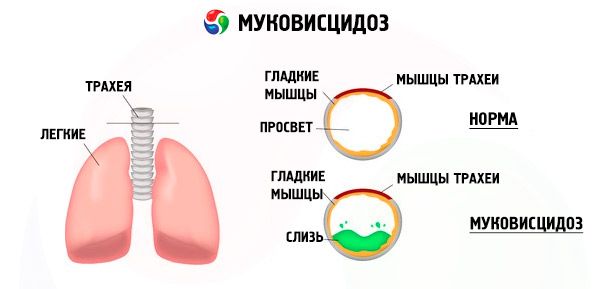
Leucócitos neutrofílicos também secretam outras enzimas proteolíticas. A alfa-1-antipirsina e o inibidor secretor de leucoproteases neutralizam a influência das enzimas proteolíticas e, portanto, protegem o sistema broncopulmonar de seus efeitos nocivos. No entanto, infelizmente, em pacientes com fibrose cística, esses fatores protetores são suprimidos por uma quantidade significativa de protease neutrofílica.
Todas essas circunstâncias contribuem para a introdução de infecção no sistema broncopulmonar e o desenvolvimento de bronquite purulenta crônica. Além disso, deve-se considerar que a proteína defeituosa codificada pelo gene da fibrose cística altera o estado funcional do epitélio brônquico, o que favorece a adesão de bactérias, principalmente Pseudomonas aeruginosa, ao epitélio brônquico.
Junto com a patologia do sistema broncopulmonar, a fibrose cística também causa danos graves ao pâncreas, estômago, intestinos grosso e delgado e fígado.
Sintomas fibrose cística
A fibrose cística se manifesta com diversos sintomas clínicos. Em recém-nascidos, a doença pode se manifestar com íleo meconial. Devido à falta ou mesmo à ausência completa de tripsina, o mecônio torna-se muito denso, viscoso e se acumula na região ileocecal. Além disso, desenvolve-se obstrução intestinal, que se manifesta por vômitos intensos com bile, distensão abdominal, ausência de excreção de mecônio, desenvolvimento de sintomas de peritonite e rápido aumento das manifestações clínicas da síndrome de intoxicação grave. A criança pode morrer nos primeiros dias de vida se não for realizada intervenção cirúrgica urgente.
Em casos menos graves, um sinal característico da fibrose cística são fezes abundantes, frequentes, untuosas, com grande quantidade de gordura e com odor muito desagradável. Em 1/3 dos pacientes, observa-se prolapso retal.
Posteriormente, os pacientes continuam a apresentar disfunção intestinal, síndrome de má absorção, distúrbios graves do desenvolvimento físico e hipovitaminose grave.
No primeiro ou segundo ano de vida, surgem sintomas de lesão do sistema broncopulmonar (forma leve da doença), que se manifestam por tosse que pode ser extremamente intensa e assemelhar-se à tosse com coqueluche. A tosse é acompanhada por cianose, falta de ar e secreção de escarro espesso, inicialmente mucoso e depois purulento. Gradualmente, desenvolve-se um quadro clínico de bronquite obstrutiva crônica e bronquiectasia, enfisema pulmonar e insuficiência respiratória. As crianças são extremamente suscetíveis a infecções respiratórias agudas virais e bacterianas, o que contribui para as exacerbações e progressão da patologia broncopulmonar. É possível o desenvolvimento de asma brônquica dependente de infecção.
Em crianças em idade escolar, a fibrose cística pode se manifestar como "cólica intestinal". Os pacientes queixam-se de dor abdominal paroxística intensa, distensão abdominal e vômitos recorrentes. À palpação do abdômen, são identificadas formações densas localizadas na projeção do intestino grosso – massas fecais misturadas com muco espesso e denso. As crianças são muito propensas ao desenvolvimento de alcalose hipoclorêmica devido à excreção excessiva de sal com o suor em climas quentes, enquanto a "geada salina" aparece na pele da criança.
Distúrbios do sistema broncopulmonar em adultos
Danos ao sistema broncopulmonar em pacientes com fibrose cística (forma pulmonar da doença) são caracterizados pelo desenvolvimento de bronquite obstrutiva purulenta crônica, bronquiectasia, pneumonia crônica, enfisema pulmonar, insuficiência respiratória e cardiopatia pulmonar. Alguns pacientes desenvolvem pneumotórax e outras complicações da fibrose cística: atelectasia, abscessos pulmonares, hemoptise, hemorragia pulmonar e asma brônquica dependente de infecção.
Os pacientes queixam-se de tosse paroxística dolorosa com expectoração mucopurulenta muito viscosa e difícil de separar, por vezes com uma mistura de sangue. Além disso, a falta de ar é extremamente característica, primeiro durante o esforço físico e depois em repouso. A falta de ar é causada por obstrução brônquica. Muitos pacientes queixam-se de rinite crônica causada por polipose e sinusite. Fraqueza significativa, diminuição progressiva do desempenho e doenças virais respiratórias agudas frequentes também são características. Ao exame, chama-se a atenção para a palidez da pele, inchaço da face, cianose das membranas mucosas visíveis e falta de ar grave. Com o desenvolvimento da cardiopatia pulmonar descompensada, surge edema nas pernas. Pode ser observado espessamento das falanges terminais dos dedos em forma de baquetas e unhas em forma de vidros de relógio. O tórax assume a forma de um barril (devido ao desenvolvimento de enfisema pulmonar).
A percussão pulmonar revela sinais de enfisema: um som de caixa, uma limitação acentuada da mobilidade da borda pulmonar e um rebaixamento da borda inferior dos pulmões. A ausculta pulmonar revela respiração ofegante com expiração prolongada, sibilância seca e dispersa e sibilância úmida, média e fina, borbulhante. No enfisema pulmonar grave, a respiração é acentuadamente prejudicada.
Manifestações extrapulmonares da fibrose cística
As manifestações extrapulmonares da fibrose cística podem ser bastante pronunciadas e ocorrer com frequência.
Danos pancreáticos
Insuficiência da função exócrina do pâncreas, de gravidade variável, é observada em 85% dos pacientes com fibrose cística. Com danos leves ao pâncreas, as síndromes de má digestão e má absorção estão ausentes, havendo apenas manifestações laboratoriais de insuficiência exócrina (baixos níveis de tripsina e lipase no sangue e no conteúdo duodenal; frequentemente, esteatorreia grave). Sabe-se que, para prevenir a síndrome de má digestão, a secreção de apenas 1 a 2% da lipase total é suficiente. Apenas distúrbios significativos da função exócrina se manifestam clinicamente.
Em condições normais, os ácinos do pâncreas produzem uma secreção líquida rica em enzimas. À medida que a secreção se move ao longo do ducto excretor, ela é enriquecida com água e ânions, tornando-se ainda mais líquida. Na fibrose cística, devido a um distúrbio na estrutura e função do regulador transmembrana (canal de cloreto), a secreção pancreática não recebe uma quantidade suficiente de líquido, torna-se viscosa e a velocidade de seu movimento ao longo do ducto excretor diminui drasticamente. As proteínas da secreção são depositadas nas paredes dos pequenos ductos excretores, resultando em sua obstrução. À medida que a doença progride, a destruição e a atrofia dos ácinos finalmente se desenvolvem - pancreatite crônica com insuficiência pancreática exócrina é formada. Isso se reflete clinicamente no desenvolvimento de síndromes de má digestão e má absorção. A insuficiência pancreática é a principal causa de má absorção de gordura na fibrose cística, mas isso geralmente é observado com uma deficiência significativa de lipase. Forsher e Durie (1991) indicam que, na ausência completa da lipase pancreática, a gordura é decomposta e absorvida em 50-60%, o que se deve à presença das lipases gástrica e salivar (sublingual), cuja atividade está próxima do limite inferior da norma. Juntamente com a interrupção da decomposição e absorção de gordura, ocorre uma interrupção da decomposição e reabsorção de proteínas. Cerca de 50% da proteína ingerida com os alimentos é perdida nas fezes. A absorção de carboidratos é afetada em menor grau, apesar da deficiência de α-amilase, mas o metabolismo de carboidratos pode ser significativamente prejudicado.
Danos ao pâncreas são expressos no desenvolvimento de síndrome de má digestão e má absorção, com perda de peso significativa e fezes gordurosas abundantes.
O desenvolvimento de síndromes de má digestão e má absorção também é facilitado pela disfunção grave das glândulas intestinais, secreção prejudicada do suco intestinal e diminuição do conteúdo de enzimas intestinais.
As síndromes de má digestão e má absorção também são chamadas de forma intestinal da fibrose cística.
A função endócrina prejudicada do pâncreas (diabetes mellitus) é observada em pacientes com fibrose cística nos estágios finais da doença (em 2% das crianças e 15% dos adultos)
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Danos ao fígado e às vias biliares
Em 13% dos pacientes com formas mistas e intestinais de fibrose cística, desenvolve-se cirrose hepática. É mais comum nas mutações W128X, delta-P508 e X1303K. A cirrose biliar hepática com hipertensão portal é detectada em 5 a 10% dos pacientes. Segundo Welch e Smith (1995), sinais clínicos, morfológicos, laboratoriais e instrumentais de lesão hepática são detectados em 86% dos pacientes com fibrose cística.
Muitos pacientes com fibrose cística também desenvolvem colecistite crônica, geralmente calculosa.
Disfunção das glândulas sexuais
Pacientes com fibrose cística podem apresentar azoospermia, que é a causa da infertilidade. A redução da fertilidade também é típica em mulheres.
Estágios
Existem três graus de gravidade da fibrose cística pulmonar.
A forma leve da fibrose cística é caracterizada por exacerbações raras (não mais do que uma vez por ano); durante os períodos de remissão, as manifestações clínicas estão praticamente ausentes e os pacientes conseguem trabalhar.
Gravidade moderada - as exacerbações ocorrem 2 a 3 vezes por ano e duram cerca de 2 meses ou mais. Na fase de exacerbação, há tosse intensa com expectoração difícil de separar, falta de ar mesmo com pequenos esforços físicos, temperatura corporal baixa, fraqueza geral e sudorese. Ao mesmo tempo, há comprometimento da função exócrina do pâncreas. Na fase de remissão, a capacidade de trabalho não é totalmente restaurada e a falta de ar durante os esforços físicos permanece.
A evolução grave é caracterizada por exacerbações muito frequentes da doença. As remissões são praticamente ausentes. O quadro clínico destaca-se por insuficiência respiratória grave, sintomas de cardiopatia pulmonar crônica, frequentemente descompensada, e hemoptise é típica. Observa-se perda de peso significativa, e os pacientes ficam completamente incapacitados. Via de regra, a patologia broncopulmonar grave é acompanhada por disfunção pancreática pronunciada.
Formulários
- Lesões broncopulmonares
- Pneumonia repetida e recorrente com curso prolongado.
- Pneumonia abscessiva, especialmente em bebês.
- Pneumonia crônica, especialmente bilateral.
- Asma brônquica refratária à terapia tradicional.
- Bronquite recorrente, bronquiolite, especialmente com cultura de Pseudomonas aeruginosa.
- Alterações no trato gastrointestinal
- Íleo meconial e seus equivalentes.
- Síndrome de absorção intestinal prejudicada de gênese desconhecida.
- Icterícia obstrutiva em recém-nascidos com curso prolongado.
- Cirrose hepática.
- Diabetes mellitus.
- Refluxo gastroesofágico.
- Colelitíase.
- Prolapso retal.
- Alterações em outros órgãos e sistemas
- Transtornos de crescimento e desenvolvimento.
- Atraso no desenvolvimento sexual.
- Infertilidade masculina.
- Pólipos nasais.
- Irmãos de famílias com fibrose cística.
[ 24 ]
Complicações e consequências
As complicações do trato gastrointestinal incluem:
- O diabetes mellitus se desenvolve em 8-12% dos pacientes com mais de 25 anos de idade.
- Colonopatia fibrosante.
- Íleo meconial no período neonatal (em 12% dos recém-nascidos com fibrose cística), síndrome de obstrução intestinal distal, prolapso retal, úlcera péptica e doença do refluxo gastroesofágico.
Complicações hepáticas:
- Doença hepática gordurosa (em 30-60% dos pacientes),
- Cirrose biliar focal, cirrose biliar multinodular e hipertensão portal associada.
A hipertensão portal às vezes leva à morte devido a varizes esofágicas.
A prevalência de colecistite e cálculos biliares é maior em pacientes com fibrose cística do que em outros indivíduos.
Puberdade tardia, diminuição da fertilidade e outras complicações. A maioria dos homens tem azoospermia e subdesenvolvimento dos canais deferentes.
Diagnósticos fibrose cística
Análise sanguínea geral - anemia de gravidade variável é típica, geralmente normo ou hipocrômica. A anemia tem gênese polifatorial (redução da absorção de ferro e vitamina B12 no intestino devido ao desenvolvimento da síndrome de má absorção). Leucopenia é possível, com o desenvolvimento de bronquite purulenta e pneumonia - leucocitose, aumento da VHS.
Análise geral de urina - sem alterações significativas, em casos raros observa-se leve proteinúria.
Exame coprológico - observam-se esteatorreia e creatorreia. Becker (1987) recomenda a dosagem de quimotripsina e ácidos graxos nas fezes. Antes da dosagem de quimotripsina nas fezes, é necessário interromper o uso de enzimas digestivas pelo menos 3 dias antes do exame. Na fibrose cística, a quantidade de quimotripsina nas fezes é reduzida e a quantidade de ácidos graxos é aumentada (a excreção normal de ácidos graxos é inferior a 20 mmol/dia). É necessário levar em consideração que o aumento da excreção de ácidos graxos nas fezes também é observado em:
- deficiência de ácidos graxos conjugados no intestino delgado devido à insuficiência hepática, obstrução dos ductos biliares, colonização bacteriana significativa do intestino delgado (neste caso, ocorre hidrólise intensiva dos ácidos biliares);
- ileíte;
- doença celíaca (com desenvolvimento de síndrome de má absorção);
- enterite;
- linfomas intestinais;
- Doença de Whipple;
- alergias alimentares;
- trânsito acelerado de massas alimentares em diarreias de várias origens, síndrome carcinoide, tireotoxicose.
Exame bioquímico de sangue - diminuição dos níveis de proteínas totais e albumina, aumento dos níveis de alfa2 e gamaglobulinas, bilirrubina e aminotransferases (em caso de lesão hepática), diminuição da atividade da amilase, lipase, tripsina e níveis de ferro e cálcio (em caso de desenvolvimento de síndrome de má digestão, má absorção).
Análise do escarro - presença de grande número de leucócitos neutrofílicos e microrganismos (durante bacterioscopia do escarro).
Um estudo da função de absorção do intestino delgado e da função exócrina do pâncreas revela distúrbios significativos.
Exame radiográfico dos pulmões - revela alterações cuja gravidade depende da gravidade e do estágio da doença. As alterações mais características são:
- aumento do padrão pulmonar devido a alterações intersticiais peribrônquicas;
- expansão das raízes dos pulmões;
- imagem de atelectasia lobular, subsegmentar ou mesmo segmentar dos pulmões;
- aumento da transparência dos campos pulmonares, principalmente nas seções superiores, posição baixa e mobilidade insuficiente do diafragma, expansão do espaço retroesternal (manifestação de enfisema pulmonar);
- infiltração segmentar ou polissegmentar do tecido pulmonar (no desenvolvimento de pneumonia).
A broncografia revela alterações causadas pela obstrução dos brônquios por escarro viscoso (fragmentação do enchimento dos brônquios com contraste, contornos irregulares, fenômeno de ruptura brônquica, diminuição significativa do número de ramos laterais), bem como broncoexaspermias (cilíndricas, mistas), localizadas principalmente nas partes inferiores dos pulmões.
A broncoscopia revela bronquite purulenta difusa com expectoração abundante, espessa e viscosa, e filmes fibrinosos.
Espirometria - já nos estágios iniciais da doença revela insuficiência respiratória do tipo obstrutiva (diminuição da CVF, VEF1, índice de Tiffno), restritiva (diminuição da CVF) ou, mais frequentemente, obstrutiva-restritiva (diminuição da CVF, CVF, VEF1, índice de Tiffno).
O teste do suor de Gibson e Cook (teste de eletrólitos no suor) envolve a estimulação da sudorese por meio da eletroforese com pilocarpina, seguida da determinação de cloretos no suor. Doerehuk (1987) descreve o teste da seguinte forma. A eletroforese com pilocarpina é realizada no antebraço, com corrente elétrica de 3 mA. Após a limpeza da pele com água destilada, o suor é coletado com papel-filtro colocado sobre a área estimulada, coberto com gaze para evitar a evaporação. Após 30 a 60 minutos, o papel-filtro é removido e eluído em água destilada. A quantidade de suor coletada é medida. Para obter resultados confiáveis, é necessário coletar pelo menos 50 mg (preferencialmente 100 mg) de suor.
Se a concentração de cloreto for superior a 60 mmol/l, o diagnóstico de fibrose cística é considerado provável; se a concentração de cloreto for superior a 100 mmol/l, o diagnóstico é confiável; neste caso, a diferença na concentração de cloro e sódio não deve exceder 8-10 mmol/l. Hadson (1983) recomenda que, se o teor de sódio e cloreto no suor estiver limítrofe, seja realizado um teste de prednisolona (5 mg por via oral durante 2 dias, seguido da determinação de eletrólitos no suor). Em indivíduos sem fibrose cística, o nível de sódio no suor diminui para o limite inferior do normal; na fibrose cística, ele não se altera. Um teste do suor é recomendado para todas as crianças com tosse crônica.
A análise de manchas de sangue ou amostras de DNA para detectar mutações importantes no gene da fibrose cística é o teste diagnóstico mais sensível e específico. No entanto, este método só é adequado para países onde a taxa de mutação delta-P508 é superior a 80%. Além disso, a técnica é muito cara e tecnicamente complexa.
O diagnóstico pré-natal da fibrose cística é realizado pela determinação das isoenzimas da fosfatase alcalina no líquido amniótico. Este método torna-se possível a partir da 18ª à 20ª semana de gestação.
Os principais critérios para o diagnóstico da fibrose cística são os seguintes:
- indicações na anamnese de atraso no desenvolvimento físico na infância, doenças respiratórias crônicas recorrentes, distúrbios dispépticos e diarreia, presença de fibrose cística em parentes próximos;
- bronquite obstrutiva crônica, frequentemente recorrente, com desenvolvimento de bronquiectasia e enfisema pulmonar, frequentemente pneumonia recorrente;
- pancreatite crônica recorrente com diminuição acentuada da função exócrina, síndrome de má absorção;
- aumento do teor de cloro no suor do paciente;
- infertilidade com função sexual preservada.
O diagnóstico bem-sucedido e o diagnóstico diferencial da fibrose cística são facilitados pela identificação de grupos de risco.
Programa de Triagem de Fibrose Cística
- Análise geral de sangue, urina, escarro.
- Análise bacteriológica do escarro.
- Análise coprológica.
- Exame bioquímico de sangue: determinação de proteínas totais e frações proteicas, glicose, bilirrubina, aminotransferases, fosfatase alcalina, gama-glutamil transpeptidases, potássio, cálcio, ferro, lipase, amilase, tripsina.
- Estudo da função exócrina do pâncreas e da função absortiva do intestino.
- Fluoroscopia e radiografia dos pulmões, tomografia computadorizada dos pulmões.
- ECG.
- Ecocardiografia.
- Broncoscopia e broncografia.
- Espirometria.
- Teste do suor.
- Consulta com um geneticista.
- Análise de manchas de sangue ou amostras de DNA para mutações importantes do gene da fibrose cística.
Quais testes são necessários?
Quem contactar?
Tratamento fibrose cística
O tipo e a gravidade dos sintomas da fibrose cística podem variar muito, por isso não há um plano de tratamento típico; ele é individualizado para cada indivíduo.
A terapia consiste nas seguintes medidas terapêuticas:
- Exercícios respiratórios e drenagem postural ajudam a eliminar o muco espesso que se acumula nos pulmões. Algumas técnicas de desobstrução das vias aéreas exigem a ajuda de familiares, amigos ou um pneumologista. Muitas pessoas usam um colete peitoral inflável que vibra em alta frequência.

- Medicamentos inalatórios que têm efeitos broncodilatadores, drenantes (mucolíticos) e antibacterianos (por exemplo, fluoroquinolonas).
- Preparações contendo enzimas pancreáticas para melhorar a digestão. Essas preparações são tomadas durante as refeições.
- Multivitaminas (incluindo vitaminas lipossolúveis).
Em 2015, a FDA aprovou um segundo medicamento para tratar a fibrose cística, que tem como alvo uma proteína defeituosa conhecida como CFTR. O primeiro medicamento, um modulador de CFTR, foi aprovado em 2012. Espera-se que os moduladores de CFTR prolonguem a vida de algumas pessoas com fibrose cística em décadas.
Pode ser necessária cirurgia para tratar as seguintes complicações respiratórias:
- Pneumotórax, hemoptise maciça recorrente ou persistente, pólipos nasais, sinusite persistente e crônica.
- Íleo meconial, intussuscepção, prolapso retal.
O transplante de pulmão é realizado no estágio terminal da doença.
Previsão
A sobrevida média dos pacientes com fibrose cística varia de 35 a 40 anos. A sobrevida média é maior em homens do que em mulheres.
Com estratégias de tratamento modernas, 80% dos pacientes chegam à idade adulta. No entanto, a fibrose cística limita significativamente as capacidades funcionais do paciente. Ainda não há cura para esta doença.